кроме макиавелли проблема метода в политических науках раннего нового времени
Димиа таб. 3мг+0.02мг №28

Показания Пероральная контрацепция. Противопоказания Противопоказания Препарат Димиа®, как и другие комбинированные пероральные контрацептивные средства (КОК), противопоказан при любом состоянии из перечисленных ниже: - тромбозы (артериальные и венозные) и тромбоэмболии в настоящее время или в анамнезе (в том числе тромбоз, тромбофлебит глубоких вен: тромбоэмболия легочной артерии, инфаркт миокарда, инсульт, цереброваскулярные нарушения). Состояния, предшествующие тромбозу (в том числе, транзиторные ишемические атаки, стенокардия) в настоящее время или в анамнезе: - множественные или выраженные факторы риска венозного или артериального тромбоза, в том числе осложненные поражения клапанного аппарата сердца, фибрилляция предсердий, заболевания сосудов головного мозга или коронарных артерий: неконтролируемая артериальная гипертензия, объёмное хирургическое вмешательство с длительной иммобилизацией, курение в возрасте старше 35 лет, ожирение с индексом массы тела >: 30 кг/м 2: - наследственная или приобретенная предрасположенность к венозному или артериальному тромбозу, например, резистентность к активированному протеину С, дефицит антитромбина III, дефицит протеина С, дефицит протеина S, гипергомоцистеинемия и антитела против фосфолипидов (наличие антител к фосфолипидам (антитела к кардиолипину, волчаночный антикоагулянт): - беременность и подозрение на нее: - период лактации: - панкреатит с выраженной гипертриглицеридемией в настоящее время или в анамнезе: - существующее тяжелое заболевание печени (или в анамнезе) при условии, что функция печени и в настоящее время не нормализована: - тяжелая хроническая или острая почечная недостаточность: - в настоящее время или в анамнезе опухоль печени (доброкачественная или злокачественная): - гормонозависимые злокачественные новообразования половых органов или молочной железы в настоящее время или в анамнезе: - кровотечение из влагалища неясного генеза: - мигрень с очаговыми неврологическими симптомами в анамнезе: - дефицит лактазы, непереносимость лактозы, глюкозо-галактозная мальабсорбция, лактазная недостаточность Лаппа: - повышенная чувствительность к препарату или любому из компонентов препарата. С осторожностью: Факторы риска развития тромбоза и тромбоэмболий: курение в возрасте до 35 лет, ожирение, дислипопротеинемия, контролируемая артериальная гипертензия, мигрень без очаговой неврологической симптоматики, неосложненные пороки клапанов сердца, наследственная предрасположенность к тромбозу (тромбозы, инфаркт миокарда или нарушение мозгового кровообращения в молодом возрасте у кого-либо из ближайших родственников): заболевания, при которых могут отмечаться нарушения периферического кровообращения: сахарный диабет без сосудистых осложнений, системная красная волчанка (СКВ), гемолитико-уремический синдром, болезнь Крона, язвенный колит, серповидно-клеточная анемия, флебит поверхностных вен: наследственный ангионевротический отек, гипертриглицеридемия, заболевания печени тяжелой степени (до нормализации функциональных проб печени): заболевания, впервые возникшие или усугубившиеся во время беременности или на фоне предыдущего приема половых гормонов (в т.ч. желтуха и/или зуд, связанные с холестазом, холелитиаз, отосклероз с ухудшением слуха, порфирия, герпес во время беременности в анамнезе, малая хорея (болезнь Сиденгама), хлоазма, послеродовый период. Беременность Препарат Димиа® противопоказан во время беременности. Если беременность наступила во время применения препарата Димиа®, его прием следует немедленно прекратить. Расширенные эпидемиологические исследования не выявили ни увеличения риска врожденных дефектов у детей, родившихся от женщин, принимавших КОК перед беременностью, ни тератогенного действия КОК при их непреднамеренном приеме во время беременности. Согласно данным доклинических исследований нельзя исключить нежелательные эффекты, оказывающие влияние на течение беременности и развитие плода ввиду гормонального действия активных компонентов. Препарат Димиа® может влиять на лактацию: уменьшать количество молока и изменять его состав. Небольшие количества контрацептивных стероидов и/или их метаболитов могут экскретироваться с молоком во время приема КОК. Эти количества могут влиять на ребенка. Применение препарата Димиа® во время грудного вскармливания противопоказано. Применение и дозы Способ применения: для прием внутрь. Как принимать препарат Димиа® Таблетки следует принимать ежедневно, примерно в одно и то же время, запивая небольшим количеством воды, в порядке, указанном на блистерной упаковке. Таблетки принимают в непрерывном режиме в течение 28 дней по 1 таблетке в сутки. Прием таблеток из следующей упаковки начинается после приема последней таблетки из предыдущей упаковки. Кровотечение "отмены" обычно начинается на 2-3 день после начала приема таблеток плацебо (последний ряд) и не обязательно заканчивается к началу следующей упаковки. Как начать прием препарата Димиа® Гормональные контрацептивы в последний месяц не использовались Прием препарата Димиа® начинается в первый день менструального цикла (т.е. в первый день менструального кровотечения). Начало приема возможно и на 2-5 день менструального цикла, в этом случае необходимо дополнительное применение барьерного метода контрацепции в течение первых 7 дней приема таблеток из первой упаковки. Переход с других комбинированных контрацептивов (комбинированные пероральные контрацептивы в виде таблеток, вагинальное кольцо или трансдермальный пластырь) Начать прием препарата Димиа® надо на следующий день после приема последней неактивной таблетки (для препаратов, содержащих 28 таблеток) или на следующий день после приема последней активной таблетки из предыдущей упаковки (возможно и на следующий день после окончания обычного 7-дневного перерыва) - для препаратов, содержащих 21 таблетку в упаковке. В случае применения женщиной вагинального кольца или трансдермального пластыря прием препарата Димиа® предпочтительно начинать в день их удаления или - самое позднее в день, когда планируется введение нового кольца или замена пластыря. Переход с контрацептивов, содержащих только прогестагены (мини-пили, инъекции, имплантаты), или с внутриматочной системы (ВМС), выделяющей прогестагены Женщина может перейти от приема мини-пили на прием препарата Димиа® в любой день (с имплантата или с ВМС в день их удаления, с инъекционных форм препаратов - в день, когда должна была быть сделана следующая инъекция), но во всех случаях необходимо использовать дополнительно барьерный метод контрацепции в течение первых 7 дней приема таблеток. После аборта в первом триместре беременности Прием препарата Димиа® может быть начат по назначению врача в день прерывания беременности. При этом женщине не нужно предпринимать дополнительные меры контрацепции. После родов или аборта во втором триместре беременности Женщине рекомендуется начинать прием препарата на 21-28 день после родов (при условии, что она не кормит грудью) или аборта во втором триместре беременности. Если прием начат позднее, женщина должна использовать дополнительно барьерный метод контрацепции в течение первых 7 дней после начала приема препарата Димиа®. С возобновлением половой жизни (до начала приема препарата Димиа®) должна быть исключена беременность. Прием пропущенных таблеток Пропуск таблетки плацебо из последнего (4-го) ряда блистера можно проигнорировать. Однако их следует выбросить во избежание непреднамеренного продления плацебо-фазы. Указания ниже относятся только к пропущенным таблеткам, содержащим действующие вещества. Если опоздание в приеме таблетки составило менее 12 ч, контрацептивная защита не снижается. Женщина должна как можно скорее принять пропущенную таблетку (как только вспомнит), а следующую таблетку - в обычное время. Если опоздание превышает 12 ч, контрацептивная защита может быть снижена. При этом можно руководствоваться двумя основными правилами: 1. Прием таблеток никогда не должен прерываться более чем на 7 дней: 2. Для достижения адекватного подавления гипоталамо-гипофизарно-яичниковой системы требуется 7 дней непрерывного приема таблеток. В соответствии с этим женщинам можно дать следующие рекомендации: - Дни 1-7 Женщина должна принять пропущенную таблетку, как только вспомнит о ней, даже если это означает прием двух таблеток одновременно. Затем она должна принимать таблетки в обычное время. Кроме того, барьерный метод, например презерватив, следует использовать в течение последующих 7 дней. Если в предшествующие 7 дней случился половой контакт, следует учитывать возможность наступления беременности. Чем больше таблеток пропущено и чем ближе этот пропуск к 7-дневному перерыву в приеме препарата, тем выше риск наступления беременности. - Дни 8-14 Женщина должна принять пропущенную таблетку, как только вспомнит об этом, даже если это означает прием двух таблеток одновременно. Затем она должна принимать таблетки в обычное время. Если в течение 7 дней, предшествующих первой пропущенной таблетке, женщина принимала таблетки как положено, необходимости в дополнительных мерах контрацепции нет. Однако если она пропустила более 1 таблетки, необходим дополнительный метод контрацепции (барьерный - например, презерватив) в течение 7 дней. - Дни 15-24 Надежность метода неизбежно снижается, поскольку приближается фаза таблеток плацебо. Однако коррекция схемы приема таблеток еще может помочь в предупреждении беременности. При выполнении одной из двух нижеописанных схем, и если в предшествующие 7 дней перед пропуском таблетки женщина соблюдала режим приема препарата, необходимость в использовании дополнительных контрацептивных мер не возникнет. Если это не так, она должна выполнить первую из двух схем и использовать дополнительные меры предосторожности в течение следующих 7 дней. 1. Женщина должна принять последнюю пропущенную таблетку сразу, как только вспомнит о ней, даже если это означает прием двух таблеток одновременно. Затем она должна принимать таблетки в обычное время до тех пор, пока активные таблетки не закончатся. 4 плацебо таблетки из последнего ряда принимать не следует, нужно сразу начать прием таблеток из следующей блистерной упаковки. Вероятнее всего, кровотечения "отмены" не будет до конца второй упаковки, но могут наблюдаться "мажущие" кровянистые выделения или кровотечение "отмены" в дни приема препарата из второй упаковки. 2. Женщина может также прервать прием активных таблеток из начатой упаковки. Вместо этого она должна принять таблетки плацебо из последнего ряда в течение 4 дней, включая дни пропуска таблеток, а затем начать прием таблеток из следующей упаковки. Если женщина пропустила прием таблеток, и впоследствии у нее не возникло кровотечение "отмены" в фазе таблеток плацебо, следует учесть возможность наступления беременности. Применение препарата при желудочно-кишечном расстройствеВ случае тяжелых желудочно-кишечных расстройств (например, рвоты или диареи) всасывание препарата будет неполным, и потребуются дополнительные меры контрацепции. Если в течение 3-4 часов после приема активной таблетки возникла рвота, необходимо как можно быстрее принять новую (замещающую) таблетку. Если возможно, следующую таблетку нужно принимать в течение 12 часов с момента обычного времени приема таблеток. Если прошло более 12 часов, рекомендуется действовать в соответствии с указаниями при пропуске таблеток. Если женщина не хочет менять обычную схему приема таблеток, она должна принять дополнительную таблетку из другой упаковки. Отсрочка менструальноподобного кровотечения "отмены" Для отсрочки кровотечения женщина должна пропустить прием таблеток плацебо из начатой упаковки и начать прием таблеток дроспиренон + этинилэстрадиол из новой упаковки. Задержку можно продлевать до тех пор, пока не закончатся активные таблетки во второй упаковке. Во время задержки у женщины могут возникнуть ациклические обильные или "мажущие" кровянистые выделения из влагалища. Регулярный прием препарата Димиа® возобновляется после фазы плацебо. Для сдвига кровотечения на другой день недели рекомендуется укоротить предстоящую фазу приема таблеток плацебо на желаемое количество дней. При укорочении цикла более вероятно, что у женщины не будет менструальноподобного кровотечения "отмены", а будут ациклические обильные или "мажущие" кровянистые выделения из влагалища при приеме следующей упаковки (так же, как при удлинении цикла). Побочные эффекты и передозировка Побочные эффекты: Во время приема препарата Димиа® зарегистрированы следующие нежелательные явления: ТАБЛИЦЪ У женщин, использующих КОК, отмечались следующие серьезные нежелательные явления: - венозные тромбоэмболические заболевания: - артериальные тромбоэмболические заболевания: - опухоли печени: - возникновение или обострение состояний, для которых связь с приемом КОК не доказана: болезнь Крона, язвенный колит, эпилепсия, мигрень, эндометриоз, миома матки, порфирия, системная красная волчанка, герпес во время предшествующей беременности, ревматическая хорея, гемолитико-уремический синдром, холестатическая желтуха: - хлоазма: - острые или хронические заболевания печени могут повлечь необходимость прекращения приема КОК до нормализации показателей функциональных проб печени: - у женщин с наследственным ангионевротическим отеком экзогенные эстрогены могут индуцировать или усиливать симптомы ангионевротического отека. Передозировка: Случаев передозировки препарата Димиа® пока не было. На основании общего опыта применения комбинированных пероральных контрацептивов потенциальными симптомами передозировки могут быть: тошнота, рвота, незначительно выраженное кровотечение из влагалища. Антидотов нет. Дальнейшее лечение должно быть симптоматическим. Взаимодействие с другими ЛС: Примечание: перед приемом сопутствующих препаратов следует прочитать инструкцию по применению препарата для выявления потенциальных взаимодействий. Влияние других лекарственных средств на препарат Димиа® Взаимодействия между пероральными контрацептивами и другим лекарственными средствами может повлечь за собой ациклическое кровотечение и/или неэффективность контрацепции. Нижеописанные взаимодействия отражены в научной литературе. Механизм взаимодействия с гидантоином, барбитуратами, примидоном, карбамазепином и рифампицином: окскарбазепином, топираматом, фелбаматом, ритонавиром, гризеофульвином и препаратами зверобоя продырявленного (hypericum perforatum) основывается на способности этих активных веществ индуцировать микросомальные ферменты печени. Максимальная индукция микросомальных ферментов печени не достигается в течение 2-3 недель, однако после этого сохраняется в течение минимум 4 недель после прекращения лекарственной терапии. Неэффективность контрацепции также отмечалась при приеме антибиотиков, например, ампициллина и тетрациклина. Механизм этого явления не ясен. Женщины при кратковременном лечении (до одной недели) любыми из вышеуказанных групп лекарственных средств или монопрепаратами должны временно использовать (в период одновременного приема других лекарственных средств и в течение еще 7 дней после его окончания), помимо КОК, барьерные методы контрацепции. Женщины, получающие терапию рифампицином, кроме приема КОК, должны использовать барьерный метод контрацепции и продолжать его применение в течение 28 дней после прекращения лечения рифампицином. Если прием сопутствующих препаратов длится дольше срока окончания активных таблеток в упаковке, прием неактивных таблеток следует прекратить и сразу начать прием таблеток дроспиренон + этинилэстрадиол из следующей упаковки. Если женщина постоянно принимает препараты-индукторы микросомальных ферментов печени, она должна использовать другие надежные негормональные методы контрацепции. Основные метаболиты дроспиренона в человеческой плазме образуются без участия системы цитохрома Р450. Ингибиторы цитохрома Р450, следовательно, вряд ли будут влиять на метаболизм дроспиренона. Влияние препарата Димиа® на другие лекарственные средства Пероральные контрацептивы могут повлиять на метаболизм некоторых других действующих веществ. Соответственно, концентрации этих веществ в плазме крови или тканях могут либо увеличиваться (например, циклоспорина), либо уменьшаться (например, ламотриджина). На основании исследований ингибирования in vitro и взаимодействий in vivo у женщин-добровольцев, принимавших омепразол, симвастатин и мидазолам в качестве субстрата, влияние дроспиренона в дозе 3 мг на метаболизм других действующих веществ маловероятно. Другие взаимодействия У пациентов без почечной недостаточности одновременный прием дроспиренона и ингибиторов ангиотензинпревращающего фермента (АПФ) или нестероидных противовоспалительных препаратов (НПВП) не оказывает значительного влияния на содержание калия в сыворотке крови. Но все же одновременное применение препарата Димиа® с антагонистами альдостерона или калийсберегающими диуретиками не исследовалось. В этом случае в ходе первого цикла лечения нужно проконтролировать концентрацию сывороточного калия. Лабораторные тесты Прием контрацептивных стероидов может повлиять на результаты некоторых лабораторных тестов, включая определение биохимических показателей функции печени, щитовидной железы, надпочечников и почек, концентрации белков плазмы (переносчиков), например кортикостероид-связывающих белков и фракций липид/липопротеин, параметров углеводного метаболизма и параметров свертывания крови и фибринолиза. В целом изменения остаются в пределах диапазона нормальных значений. Дроспиренон является причиной увеличения активности ренина в плазме крови и - за счет небольшой атиминералокортикоидной активности - снижает концентрацию альдостерона в плазме. Фармакологическое действие и фармакокинетика Препарат Димиа® является комбинированным монофазным пероральным контрацептивным средством (КОК), содержащим дроспиренон и этинилэстрадиол. По своему фармакологическому профилю дроспиренон близок к натуральному прогестерону: не обладает эстрогенной, глюкокортикоидной и антиглюкокортикоидной активностью и характеризуется выраженным антиандрогенным и умеренным антиминералокортикоидным действием. Контрацептивный эффект основывается на взаимодействии различных факторов, важнейшими из которых являются торможение овуляции, увеличение вязкости секрета шейки матки и изменение эндометрия. Индекс Перля, показатель, отражающий частоту наступления беременности у 100 женщин репродуктивного возраста в течение года применения контрацептива, составляет менее 1. Фармакокинетика: Дроспиренон Всасывание При пероральном приеме дроспиренон быстро и практически полностью всасывается в желудочно-кишечном тракте. Максимальные концентрации дроспиренона в сыворотке - около 38 нг/мл - достигаются примерно через 1-2 ч после однократного приема. Биодоступность 76-85%. Одновременный прием с пищей не влияет на биодоступность дроспиренона. Распределение После приема внутрь концентрации дроспиренона в плазме крови уменьшались с конечным периодом полувыведения 31 ч. Дроспиренон связывается с сывороточным альбумином и не связывается с глобулином, связывающим половые гормоны (ГСПГ) или с кортикостероид-связывающим глобулином (транскортин). Всего 3-5% общей сывороточной концентрации дроспиренона существует в виде свободных стероидов. Индуцированное этинилэстрадиолом увеличение ГСПГ не влияет на связывание дроспиренона с сывороточными белками. Средний кажущийся объем распределения дроспиренона составляет 3,7 ± 1,2 л/кг. Метаболизм Дроспиренон активно метаболизируется после приема внутрь. Главные метаболиты в плазме крови - это кислотные формы дроспиренона, образовавшиеся при раскрытии лактонного кольца, и 4,5-дигидро-дроспиренон-З-сульфат, оба образуются без участия системы Р450. Дроспиренон в малой степени метаболизируется цитохромом Р450 3А4 и способен ингибировать этот фермент, а также цитохром Р450 1А1, цитохром Р450 2С9 и цитохром Р450 2С19 in vitro. Выведение Почечный клиренс метаболитов дроспиренона в сыворотке крови составляет 1,5 ± 0,2 мл/мин/кг. Дроспиренон экскретируется только в следовых количествах в неизмененном виде. Метаболиты дроспиренона выводятся почками и через кишечник с соотношением экскреции около 1,2:1,4. Период полувыведения метаболитов почками и через кишечник составляет около 40 ч. Равновесная концентрация В ходе цикла лечения максимальная равновесная концентрация дроспиренона в плазме крови составляет около 70 нг/мл, она достигается после 8 дней лечения. Сывороточные концентрации дроспиренона увеличиваются примерно в 3 раза вследствие соотношения конечного периода полувыведения и интервала дозирования. Этинилэстрадиол Всасывание При пероральном приеме этинилэстрадиол всасывается быстро и полностью. Максимальная концентрация в сыворотке крови - около 33 пкг/мл - достигается в течение 1-2 часов после однократного приема внутрь. Абсолютная биодоступность в результате пресистемной конъюгации и пресистемного метаболизма составляет приблизительно 60%. Одновременный прием пищи уменьшал биодоступность этинилэстрадиола примерно у 25% обследованных пациенток: у других изменений не было. Распределение Сывороточные концентрации этинилэстрадиола уменьшались двухфазно, в фазе конечного распределения период полувыведения составляет примерно 24 часа. Этинилэстрадиол хорошо, но неспецифически связывается с сывороточным альбумином (примерно 98,5%) и индуцирует увеличение концентраций в сыворотке крови ГСПГ. Кажущийся объем распределения - около 5 л/кг. Метаболизм Этинилэстрадиол является субстратом пресистемной конъюгации в слизистой оболочке тонкого кишечника и в печени. Этинилэстрадиол первично метаболизируется путем ароматического гидроксилирования, при этом образуется широкий ряд гидроксилированных и метилированных метаболитов, которые присутствуют как в свободной форме, так и в виде конъюгатов с глюкуроновой кислотой. Почечный клиренс метаболитов этинилэстрадиола составляет примерно 5 мл/мин/кг. Выведение Неизмененный этинилэстрадиол практически не выводится из организма. Метаболиты этинилэстрадиола выводятся почками и через кишечник в соотношении 4:6. Период полувыведения метаболита составляет около 24 часов. Равновесная концентрация Равновесная концентрация наступает во второй половине цикла лечения, а сывороточная концентрация этинилэстрадиола увеличивается в 2,0-2,3 раза. Фармакокинетика у особых групп пациентов При нарушении функции почек Равновесная концентрация дроспиренона в плазме крови у женщин с почечной недостаточностью легкой степени тяжести (клиренс креатинина (КК) 50-80 мл/мин) была сравнима с соответствующими показателями у женщин с нормальной функцией почек (КК >: 80 мл/мин). У женщин с почечной недостаточностью средней степени тяжести (КК от 30 мл/мин до 50 мл/мин) концентрация дроспиренона в плазме крови была в среднем на 37% выше, чем у женщин с нормальной функцией почек. Дроспиренон хорошо переносился во всех группах. Прием дроспиренона не оказывал клинически значимого влияния на содержание калия в сыворотке крови. Фармакокинетика при тяжелой почечной недостаточности не изучалась. При нарушении функции печени Дроспиренон хорошо переносится пациентами с легкой и умеренной степенью печеночной недостаточности (класс В по Чайлд-Пью). Фармакокинетика при тяжелой печеночной недостаточности не изучалась. Особые указания Если есть любые состояния/факторы риска из числа упомянутых ниже, пользу от приема КОК следует оценивать индивидуально для каждой женщины и обсуждать с ней перед началом применения. При обострении нежелательного явления или в случае появления любого из этих состояний или факторов риска же нщина должна связаться с лечащим врачом. Врач должен решить, следует ли прервать прием КОК. Нарушения кровообращения Прием любого комбинированного перорального контрацептива увеличивает риск венозной тромбоэмболии (ВТЭ). Повышение риска ВТЭ наиболее выражено на первом году применения женщиной комбинированного перорального контрацептива. Эпидемиологические исследования показали, что частота ВТЭ у женщин с отсутствием факторов риска, принимавших низкие дозы эстрогенов (<: 0,05 мг этинилэстрадиола) в составе комбинированного перорального контрацептива, составляет примерно 20 случаев на 100000 женщин-лет (для левоноргестрелсодержащих КОК “второго поколения”) или 40 случаев на 100000 женщин-лет (для дезогестрел/гестоденсодержащих КОК “третьего поколения”). У женщин, не пользующихся КОК, случается 5-10 ВТЭ и 60 беременностей на 100000 женщин-лет. ВТЭ фатальна в 1-2% случаев. Данные крупного проспективного с 3-направлениями исследования показали, что частота ВТЭ у женщин с другими факторами риска венозной тромбоэмболии или без них, применявших комбинацию этинилэстрадиола и дроспиренона, 0,03 мг + 3 мг, совпадает с частотой ВТЭ у женщин, применявших левоноргестрелсодержащие пероральные контрацептивы и другие КОК. Степень риска венозной тромбоэмболии при приеме препарата Димиа® в настоящее время не установлена. Эпидемиологические исследования также выявили связь приема КОК с увеличением риска артериальной тромбоэмболии (инфаркт миокарда, транзиторные ишемические нарушения). Очень редко у женщин, принимающих пероральные контрацептивы, происходил тромбоз других кровеносных сосудов, например вен и артерий печени, брыжейки, почек, головного мозга или сетчатки. Единого мнения относительно связи этих явлений с приемом гормональных контрацептивов нет. Симптомы венозных или артериальных тромботических/тромбоэмболических явлений или острых нарушений мозгового кровообращения: - необычная односторонняя боль и/или отечность нижних конечностей: - внезапная сильная боль в груди, вне зависимости от того, отдает ли она в левую руку или нет: - внезапная одышка: - внезапное появление кашля: - любая необычная тяжелая длительная головная боль: - внезапная частичная или полная потеря зрения: - диплопия: - нарушенная речь или афазия: - вертиго: - коллапс с парциальными эпилептическими припадками или без них: - слабость или очень заметное онемение, внезапно поразившее одну сторону или одну часть тела: - двигательные расстройства: - "острый" живот. Перед началом приема КОК женщине следует проконсультироваться со специалистом. Риск венозных тромбоэмболических нарушений при приеме комбинированных пероральных контрацептивов (КОК) возрастает при: - увеличении возраста: - наследственной предрасположенности (венозная тромбоэмболия случалась когда- либо у братьев-сестер или родителей в относительно раннем возрасте): - длительной иммобилизации, расширенном оперативном вмешательстве, любом хирургическом вмешательстве на нижних конечностях или крупной травме. В таких ситуациях рекомендуется прекратить прием препарата (в случае планового хирургического вмешательства минимум за четыре недели) и не возобновлять до истечения двух недель после полного восстановления подвижности. Если прием препарата не был прекращен заблаговременно, следует рассмотреть возможность антикоагулянтного лечения: - ожирении (индекс массы тела более 30 кг/м 2): - отсутствии единого мнения о возможной роли варикоза вен и поверхностного тромбофлебита при появлении или обострении венозного тромбоза. Риск артериальных тромбоэмболических осложнений или острого нарушения мозгового кровообращения при приеме комбинированных пероральных контрацептивов (КОК) возрастает при: - увеличении возраста: - курении (женщинам старше 35 лет настоятельно рекомендуется бросить курить, если они хотят принимать КОК): - дислипопротеинемии: - артериальной гипертензии: - мигрени без очаговой неврологической симптоматики: - ожирении (индекс массы тела более 30 кг/м 2): - наследственной предрасположенности (артериальная тромбоэмболия когда-либо у братьев-сестер или родителей в относительно раннем возрасте). Если наследственная предрасположенность возможна, женщине следует проконсультироваться со специалистом перед началом приема КОК. поражении клапанов сердца: фибрилляции предсердий. Наличие одного серьезного фактора риска заболевания вен или нескольких факторов риска заболевания артерий также может являться противопоказанием. Также следует рассмотреть возможность антикоагулянтной терапии. Женщины, принимающие КОК, должны быть должным образом инструктированы о необходимости информирования лечащего врача в случае возникновения подозрений на симптомы тромбоза. В случае, если тромбоз заподозрен или подтвержден, прием КОК следует прекратить. Нужно начать адекватную альтернативную контрацепцию в силу тератогенности антикоагулянтной терапии (непрямыми антикоагулянтами - производными кумарина). Следует учитывать увеличенный риск развития тромбоэмболии в послеродовом периоде. Другие медицинские состояния, связанные с нежелательными сосудистыми явлениями, включают сахарный диабет, системную красную волчанку, гемолитико-уремический синдром, хроническое воспалительное заболевание кишечника (болезнь Крона или язвенный колит) и серповидно-клеточную анемию. Увеличение частоты или тяжести мигрени на фоне приема КОК может быть показанием к немедленной отмене комбинированных пероральных контрацептивов. Опухоли Самым значительным фактором риска развития рака шейки матки является инфицирование вирусом папилломы человека. В некоторых эпидемиологических исследованиях сообщалось о повышенном риске развития рака шейки матки при длительном применении комбинированных пероральных контрацептивов, однако сохраняются противоречивые мнения относительно того, в какой степени эти находки относятся к сопутствующим факторам, например, исследованию на наличие рака шейки матки или применению барьерных методов контрацепции. Мета-анализ результатов 54 эпидемиологических исследований выявил небольшое увеличение относительного риска (RR = 1,24) рака молочной железы у женщин, которые в настоящее время принимают КОК. Риск постепенно снижается в течение 10 лет после прекращения приема КОК. Так как рак молочной железы редко развивается у женщин до 40 лет, увеличение числа диагностированных случаев рака молочной железы у применяющих КОК мало влияет на общую вероятность возникновения рака молочной железы. В этих исследованиях не было выявлено достаточных доказательств причинно- следственной связи. Повышение риска может быть следствием более ранней диагностики рака молочной железы у применяющих КОК, биологического действия КОК или комбинации обоих факторов. Диагностированный рак молочной железы у женщин, когда- либо принимавших КОК, клинически был менее тяжелым, что обусловлено ранней диагностикой заболевания. Редко у женщин, принимавших КОК, возникали доброкачественные опухоли печени и, еще более редко, злокачественные опухоли печени. В отдельных случаях эти опухоли были угрожающими жизни из-за внутрибрюшного кровотечения. Это следует учитывать при проведении дифференциального диагноза в случае появления сильных болей в области живота, увеличения печени или признаков внутрибрюшного кровотечения. Другие Прогестагеновый компонент препарата Димиа® - это антагонист альдостерона, удерживающий калий в организме. В большинстве случаев увеличение содержание калия не ожидается. Однако в клиническом исследовании у некоторых пациентов с легкими или умеренными заболеваниями почек, которые принимали калийсберегающие препараты, содержание калия в сыворотке незначительно возрастает во время приема дроспиренона. Следовательно, рекомендуется контролировать содержание калия в сыворотке во время первого цикла лечения у пациенток с почечной недостаточностью, у которых концентрация калия в сыворотке до лечения находилась на уровне верхней границы нормы и, особенно, при одновременном приеме калийсберегающих препаратов. У женщин с гипертриглицеридемией или наследственной предрасположенностью к таковой может быть увеличен риск возникновения панкреатита при приеме КОК. Хотя небольшое повышение артериального давления отмечалось у многих женщин, принимавших комбинированные пероральные контрацептивы (КОК), клинически значимое повышение случалось редко. Только в этих редких случаях обоснованно немедленное прекращение приема КОК. Если при приеме КОК у пациенток с сопутствующей артериальной гипертензией постоянно увеличивается артериальное давление (АД), или значительно повышенное давление нельзя скорректировать антигипертензивными препаратами, прием КОК следует прекратить. После нормализации АД с помощью антигипертензивных препаратов прием КОК можно возобновить. Нижеперечисленные заболевания появлялись или обострялись и при беременности, и при приеме КОК, однако доказательства их взаимосвязи с приемом КОК неубедительны: желтуха и/или зуд, связанный с холестазом, камни в желчном пузыре: порфирия: системная красная волчанка: гемолитико-уремический синдром: ревматическая хорея (хорея Сиденгама): герпес при беременности: отосклероз с потерей слуха. У женщин с наследственным ангионевротическим отеком экзогенные эстрогены могут индуцировать или усиливать симптомы отека. Острые или хронические заболевания печени могут быть показанием к прекращению приема КОК до нормализации показателей функции печени. Рецидив холестати
1007 Руб.
Светодиодная LED маска для омоложения кожи лица и шеи с 7 цветами Gezatone m1030

Маска для светотерапии с семью цветами светового спектра оказывает многогранное влияние, корректируя косметические проблемы кожи лица и шеи, замедляет возрастные изменения, улучшает цвет и внешний вид кожи.ЛЕД маска для лица m 1030 Жезатон обладает большими возможностями, ведь в ней сочетаются 7 цветов, которые решают проблемы разных типов кожи. Кроме того, воздействие световыми волнами разной длины усиливает действие корректирующей косметики, доставляя активные вещества точно по назначению. Применение беспроводной светодиодной маски рекомендуется для омоложения, профилактики раннего старения и оказания корректирующего косметического действия. На выбор цвета влияет исходное состояние и тип кожных покровов. M1030 Gezatone рекомендуется при проблемах:Возрастные признаки, атония и потеря тургора, формирование заломов, морщины разной локализации, снижение эластических свойств. Проблема дефицита влаги, сухость, дискомфорт и сопровождающие дегидратацию мелкие морщинки Светотерапия эффективна для любого типа, при «коже курильщика», бледности, стрессовой и уставшей коже. Повышенная продукция себума, несовершенства и воспаления, акне и постакне, расширенные поры и сниженный тонус кожных покровов. Изменение цвета, тусклая кожа, проявление пигментации, неровности микрорельефа в виде небольших шрамов и рубчиков. При повышенной кожной реактивности и склонности к красноте, лед маска снижает дискомфортные ощущения и эритему. Светодиодная led маска восстанавливает кожные покровы после стрессовых и травмирующих влияний, помогает подготовить кожу к процедурам и сократить восстановительный период. Также, светодиодная маска для омоложения лица m 1030 поможет предотвратить ранее старение и применяется для профилактических сеансов. На глубину доставки косметического средства влияет длина волны: чем длиннее волна, тем глубже проникнут активные компоненты препарата. Этот процесс носит название «фотопорации». При использовании светодиодной маски для омоложения m 1030 необходимо использовать специальнуюсыворотку или коллагеновую, гидрогелевую, тканевую маску, действие которой многократно усилится под влиянием световых волн. Результаты применения светодиодной led маски для лица m 1030 Жезатон. Сеансы светотерапии освежают цвет лица и улучшают состояние кожных покровов. Улучшается уровень гидратации, сокращаются линии обезвоженности, уходят дискомфортные ощущения. Кожа спокойная и гладкая. Сокращаются признаки возраста, морщинки постепенно разглаживаются, кожа становится более упругой. Уменьшается пигментация и выравнивается тон кожных покровов. При воздействии на проблемную кожу, уменьшается воспаление, сужаются поры, сокращается продукция себума. Снижается частота рецидивов воспалений до 74%. Чувствительная кожа становится менее раздражимой, повышается её невосприимчивость к стрессорам. Поврежденная окружающей средой или косметическими манипуляциями кожа быстрее восстанавливается, избавляется от отеков и красноты. «Уставшая» кожа обретает прежнюю яркость, становится упругой, увлажненной и эластичной. Сыворотка помогает снизить реактивность кожи и повысить сопротивляемость кожи раздражающим факторам.5 причин купить светодиодную маску для лица m 1030. Комфортные процедуры, которые не занимают много времени, помогут сберечь бюджет, ведь стоимость лед маски gezatone сравнима с двумя сеансами в салоне. Сеансы светотерапии можно проводить в любом возрасте, 16 . Обладатели разных типов кожи, применяя маску Жезатон, избавятся от косметических проблем, присущих коже их типа. Светотерапия безопасна, не разогревает и не травмирует кожу, она не повышает чувствительность к солнечным лучам. В приборе не используется ультрафиолет. Маску можно применять в период активного загара. Беспроводная конструкция позволяет использовать этот бьюти-девайс в любом удобном месте, без поиска электрической розетки. Смарт-таймер самостоятельно выключает маску через 10 минут, не нужно следить за часами и контролировать время сеанса. Светотерапия с маской gezatone усиливает действие косметики до 60%, может проводиться при гиперчувствительной коже и при куперозе, в то время, когда многие аппаратные методы противопоказаны. Противопоказания. Беременность. Прием препаратов для лечения акне в течение последнего полугодового периода или прием лекарственных средств, повышающих фоточувствительность. Онкология, аутоиммунные патологии кожных покровов, фотодерматозы. Повреждения и травмы кожных покровов Эпилепсия, психические заболевания Повышенная температура, простуда, грипп. При наличии в анамнезе гипертонической болезни, сердечно-сосудистых заболеваний и кардиостимулятора, рекомендуется проконсультироваться с врачом. Способ применения. Зарядите аккумулятор. Перед сеансом удалите косметику и нанесите на кожу специальную сыворотку для LED терапии или маску на основе коллагена или гидрогеля. Наденьте маску и включите, выберите нужный цвет, последовательно нажимая на кнопку питания. Через 10 минут прибор выключит смарт-таймер. Завершите сеанс кремом по типу кожи. Комплектация. Комплектация Светодиодная маска m 1030 Gezatone – 1 шт. Кабель USB – 1 шт. Инструкция по эксплуатации – 1 шт
2700 Руб.Галвус Мет таб. п/о 50мг+850мг №30
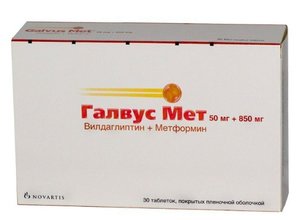
Показания Сахарный диабет 2 типа (в сочетании с диетотерапией и физическими упражнениями): - при недостаточной эффективности монотерапии вилдаглиптином или метформином: - у пациентов, ранее получающих комбинированную терапию вилдаглиптином и метформином в виде монопрепаратов: - в комбинации с производными сульфонилмочевины (тройная комбинированная терапия) у пациентов, ранее получавших терапию производными сульфонилмочевины и метформином без достижения адекватного контроля гликемии: - в тройной комбинированной терапии с инсулином у пациентов, ранее получавших инсулинотерапию в стабильной дозе и метформин без достижения адекватного контроля гликемии: - в качестве начальной терапии у пациентов с сахарным диабетом 2 типа при недостаточной эффективности диетотерапии, физических упражнений и при необходимости улучшения контроля гликемии. Противопоказания Противопоказания - Повышенная чувствительность к вилдаглиптину или метформину или любым другим компонентам препарата: - почечная недостаточность или нарушения функции почек (при концентрации креатинина в сыворотке крови 1,5 мг% (>:135 мкмол/л) для мужчин и 1,4 мг% (>:110 мкмол/л) для женщин): - острые состояния, с риском развития нарушения функции почек: дегидратация (при диарее, рвоте), лихорадка, тяжелые инфекционные заболевания, состояния гипоксии (шок, сепсис, инфекции почек, бронхолегочные заболевания): - острая и хроническая сердечная недостаточность, острый инфаркт миокарда, острая сердечно-сосудистая недостаточность (шок), дыхательная недостаточность: - нарушения функции печени: - острый или хронический метаболический ацидоз (включая диабетический кетоацидоз в сочетании с комой или без таковой). Диабетический кетоацидоз должен корректироваться инсулинотерапией. Лактоацидоз (в том числе и в анамнезе): - препарат не следует применять за 48 часов перед хирургическими операциями, радиоизотопными, рентгенологическими исследованиями с введением контрастных средств и в течение 48 часов после их проведения: - беременность и период грудного вскармливания: - сахарный диабет 1 типа: - хронический алкоголизм, острое отравление алкоголем: - соблюдение низкокалорийной диеты (менее 1000 ккал/сут): Эффективность и безопасность применения препарата у детей до 18 лет не установлена. Поскольку у пациентов с нарушением функции печени в ряде случаев отмечался лактоацидоз, возможно, являющийся одним из побочных эффектов метформина, препарат Галвус Мет не следует применять у пациентов с заболеваниями печени или нарушениями биохимических показателей функции печени. С осторожностью: Препараты, содержащие метформин, рекомендуется применять с осторожностью у пациентов старше 60 лет при выполнении тяжелой физической работы, в связи с повышенным риском развития у них лактоацидоза. Беременность Беременность В экспериментальных исследованиях у животных при применении вилдаглиптина в дозах, в 200 раз превышающих рекомендуемые, препарат не вызывал нарушения раннего развития эмбриона и не оказывал тератогенного действия. При применении вилдаглиптина в комбинации с метформином в соотношении 1:10 также не было выявлено тератогенного действия. Поскольку достаточных данных по применению препарата Галвус Мет при беременности нет, применение препарата при беременности противопоказано. Грудное вскармливание Метформин проникает в грудное молоко. Неизвестно, выделяется ли вилдаглиптин с грудным молоком. Применение препарата Галвус Мет в период грудного вскармливания противопоказано. Применение и дозы Препарат применяют внутрь. Режим дозирования препарата Галвус Мет следует подбирать индивидуально в зависимости от эффективности и переносимости терапии. При применении препарата Галвус Мет не следует превышать рекомендованную максимальную суточную дозу вилдаглиптина (100 мг). Рекомендуемую начальную дозу препарата Галвус Мет следует подбирать, учитывая длительность течения сахарного диабета и уровень гликемии, состояние пациента и уже применявшиеся у пациента схемы лечения вилдаглиптином и/или метформином. Для уменьшения выраженности побочных эффектов со стороны органов желудочно-кишечного тракта, характерных для метформина, препарат Галвус Мет принимают во время еды. Начальная доза препарата Галвус Мет при неэффективности монотерапии вилдаглиптином Лечение препаратом Галвус Мет можно начинать с одной таблетки дозировкой 50 мг+500 мг 2 раза в сутки: после оценки терапевтического эффекта дозу можно постепенно увеличивать. Начальная доза препарата Галвус Мет при неэффективности монотерапии метформином В зависимости от дозы уже принимаемого метформина, лечение препаратом Галвус Мет можно начинать с одной таблетки дозировкой 50 мг+500 мг, 50 мг+850 мг или 50 мг+1000 мг 2 раза в сутки. Начальная доза препарата Галвус Мет у пациентов, ранее получавших комбинированную терапию вилдаглиптином и метформином в виде отдельных таблеток В зависимости от доз уже принимаемых вилдаглиптина или метформина, лечение препаратом Галвус Мет следует начинать с таблетки, максимально близкой по дозировке к существующему лечению, 50 мг+500 мг, 50 мг+850 мг или 50 мг+1000 мг, и корректировать дозу в зависимости от эффективности. Стартовая доза препарата Галвус Мет в качестве начальной терапии у пациентов с сахарным диабетом 2 типа при недостаточной эффективности диетотерапии и физических упражнений В качестве стартовой терапии препарат Галвус Мет следует применять в начальной дозе 50 мг+500 мг однократно в сутки и после оценки терапевтического эффекта постепенно увеличивать дозу до 50 мг+1000 мг дважды в сутки. Комбинированная терапия препаратом Галвус Мет и производными сульфонилмочевины или инсулином Доза препарата Галвус Мет рассчитывается, исходя из дозы вилдаглиптина 50 мг х 2 раза в день (100 мг в день) и метформина в дозе, равной принимаемой ранее в виде монопрепарата. Пациенты с нарушением функции почек У пациентов с нарушением функции почек может потребоваться коррекция дозы препарата при клиренсе креатинина (КК, рассчитанном по формуле Кокрофта-Голта) в диапазоне от 60 до 90 мл/мин. Применение препарата Галвус Мет у пациентов с КК <:60 мл/мин противопоказано. Применение у пациентов в возрасте 65 лет Метформин выводится почками. Поскольку у пациентов старше 65 лет часто отмечается нарушение функции почек, дозу препарата Галвус Мет у данных пациентов следует корректировать, основываясь на показателях функции почек. При применении препарата у пациентов старше 65 лет необходимо регулярно контролировать функцию почек. Применение у пациентов в возрасте младше 18 лет Поскольку безопасность и эффективность препарата Галвус Мет у детей и подростков младше 18 лет не изучена, применение препарата противопоказано у данной категории пациентов. Побочные эффекты и передозировка Побочные эффекты: Представленные ниже данные относятся к применению вилдаглиптина и метформина в монотерапии и в комбинации. На фоне терапии вилдаглиптином редко отмечались нарушения функции печени (включая гепатит) бессимптомного течения. В большинстве случаев данные нарушения и отклонения показателей функции печени от нормы разрешались самостоятельно без осложнений после прекращения терапии препаратом. При применении вилдаглиптина в дозе 50 мг 1 или 2 раза в сутки частота повышения активности "печеночных" ферментов (аланинаминотрансферазы (АЛТ) или аспартатаминотрансферазы (ACT) в 3 раза выше верхней границы нормы (ВГН)) составляла 0,2% или 0,3% соответственно (по сравнению с 0,2% в контрольной группе). Повышение активности "печеночных" ферментов в большинстве случаев было бессимптомным, не прогрессировало и не сопровождалось холестазом или желтухой. Для оценки частоты развития нежелательных явлений (НЯ) использовались следующие критерии: очень часто ( 1/10), часто ( 1/100, <:1/10), нечасто ( 1/1000, <:1/100), редко ( 1/10,000,<:1/1,000), очень редко (<:1/10,000), включая отдельные случаи. Нежелательные реакции, возможно связанные с применением комбинированной терапии вилдаглиптином и метформином (частота развития которых в группе вилдаглиптина и метформина отличалась от таковой на фоне применения плацебо и метформина более чем на 2%) представлены ниже. Нарушения со стороны нервной системы: часто - головная боль, головокружение, тремор. При применении вилдаглиптина в комбинации с метформином в различных дозах гипогликемия отмечалась в 0,9% случаев (для сравнения в группе плацебо в комбинации с метформином - в 0,4%). Частота НЯ со стороны желудочно- кишечного тракта на фоне комбинированной терапии вилдаглиптином и метформином составляла 12,9%. При применении метформина подобные НЯ отмечались у 18,1% пациентов.В группах пациентов, получавших метформин в комбинации с вилдаглиптином, нарушения со стороны желудочно-кишечного тракта отмечались с частотой 10-15%, а в группе пациентов, получавших метформин в комбинации с плацебо, - с частотой 18%. Долгосрочные клинические исследования продолжительностью до 2 лет не выявили каких-либо дополнительных отклонений профиля безопасности или непредвиденных рисков при применении вилдаглиптина в монотерапии. Изучение применения комбинации вилдаглиптина и метформина в качестве стартовой терапии при сахарном диабете 2 типа не выявило рисков и дополнительных данных по безопасности применения. При применении вилдаглиптина одновременно с инсулином В контролируемых клинических исследованиях при применении вилдаглиптина в дозе 50 мг 2 раза в день в комбинации с инсулином в сочетании с метформином или без него, частота отмены терапии в связи с развитием побочных реакций составила 0,3% в группе вилдаглиптина, при этом в группе плацебо случаев отмены терапии не было. Частота гипогликемии была сопоставимой в обеих группах (14,0% в группе вилдаглиптина и 16,4% в группе плацебо).В группе вилдаглиптина отмечены случаи гипогликемии тяжелой степени у двух пациентов, в группе плацебо - у 6 пациентов. На момент завершения исследования вилдаглиптин не оказывал влияния на среднюю массу тела (масса тела увеличена на +0,6 кг по сравнению с исходной в группе вилдаглиптина, в группе плацебо изменений не отмечено).НЯ у пациентов, получавших вилдаглиптин 50 мг 2 раза в день в комбинации с инсулином (с метформином или без него), представлены ниже. Нарушения со стороны нервной системы: часто - головная боль. Нарушения со стороны желудочно-кишечного тракта: часто - тошнота, гастроэзофагеальный рефлюкс: нечасто - диарея, метеоризм. Нарушения со стороны обмена веществ и питания: часто - гипогликемия. Общие расстройства и нарушения в месте введения: часто - озноб. При применении вилдаглиптина в комбинации с препаратами сульфонилмочевины. Случаев отмены препарата, связанных с развитием НЯ в группе комбинированной терапии вилдаглиптином, метформином и глимепиридом, отмечено не было. В группе комбинированной терапии плацебо, метформином и глимепиридом частота НЯ составила 0,6%. Гипогликемия отмечалась часто в обеих группах (5,1% в группе комбинированной терапии вилдаглиптином, метформином и глимепиридом и 1,9% в группе комбинированной терапии плацебо, метформином и глимепиридом). В группе вилдаглиптина отмечен один эпизод гипогликемии тяжелой степени. На момент завершения исследования значимого влияния на массу тела выявлено не было (+0,6 кг в группе вилдаглиптина и -0,1 кг в группе плацебо). НЯ у пациентов, получавших вилдаглиптин 50 мг 1 или 2 раза в день в комбинации препаратами сульфонилмочевины, представлены ниже (с метформином или без него). Инфекционные и паразитарные заболевания: очень редко - назофарингит. Нарушения со стороны нервной системы: часто - головокружение, тремор, астения. Нарушения со стороны желудочно-кишечного тракта: нечасто - запор. Нарушения со стороны обмена веществ и питания: часто - гипогликемия. Нарушения со стороны кожи и подкожных тканей: часто - гипергидроз. При применении вилдаглиптина в монотерапии. Инфекционные и паразитарные заболевания: очень редко - инфекции верхних дыхательных путей, назофарингит. Нарушения со стороны нервной системы: часто - головокружение: нечасто головная боль. Нарушения со стороны желудочно-кишечного тракта: нечасто - запор. Нарушения со стороны кожи и подкожных тканей: нечасто - кожная сыпь. Нарушения со стороны скелетно-мышечной и соединительной ткани: часто - артралгия. Нарушения со стороны сосудов: нечасто - периферические отеки. При применении комбинированной терапии вилдаглиптином и метформином не отмечалось клинически значимого повышения частоты вышеуказанных НЯ, отмечавшихся при приеме вилдаглиптина. На фоне монотерапии вилдаглиптином или метформином частота развития гипогликемии составляла 0,4% (нечасто). Монотерапия вилдаглиптином и комбинированное лечение вилдаглиптин+метформин не оказывали влияния на массу тела пациентов. Долгосрочные клинические исследования продолжительностью до 2 лет не выявили каких-либо дополнительных отклонений профиля безопасности или непредвиденных рисков при применении вилдаглиптина в монотерапии. Пострегистрационные исследования В пострегистрационном периоде выявлены следующие побочные реакции (поскольку данные сообщаются в добровольном порядке от популяции неопределенного размера, достоверно определить частоту развития данных НЯ не представляется возможным, в связи с чем они классифицированы как частота неизвестна): гепатит (обратим при прекращении терапии), повышение активности "печеночных" ферментов, миалгия, крапивница, панкреатит, буллезное и эксфолиативное поражения кожи, артралгия, в редких случаях выраженная. При применении метформина в монотерапии. Нарушения со стороны обмена веществ и питания: очень часто - снижение аппетита: очень редко - лактоацидоз. Нарушения со стороны желудочно-кишечного тракта: очень часто - метеоризм, тошнота, рвота, диарея, боль в животе: часто - дисгевзия. Нарушения со стороны печени и желчевыводящих путей: очень редко - гепатит. Нарушения со стороны кожи и подкожных тканей: очень редко - кожные реакции (в частности, эритема, зуд, крапивница). Лабораторные и инструментальные данные: очень редко - уменьшение всасывания витамина В12, изменение показателей функции печени. Уменьшение всасывания витамина В12 и снижение его концентрации в сыворотке крови на фоне применения метформина отмечалось очень редко у пациентов, получавших препарат в течение длительного времени, и, как правило, не представляло клинического значения. Следует учитывать возможность уменьшения всасывания витамина В12 у пациентов с мегалобластной анемией. Отдельные случаи гепатита, которые наблюдались на фоне применения метформина, разрешались после его отмены. Если отмечено ухудшение типического течения любого из указанных в инструкции побочных эффектов или Вы заметили любые другие побочные эффекты, не указанные в инструкции, сообщите об этом врачу. Передозировка: Вилдаглиптин Вилдаглиптин хорошо переносится в дозе до 200 мг/сут. При применении препарата в дозе 400 мг/сут могут наблюдаться боли в мышцах, редко - легкие транзиторные парестезии, лихорадка, отеки и транзиторное повышение активности липазы (выше ВГН в 2 раза). При увеличении лозы вилдаглиптина до 600 мг/сут возможно развитие отеков конечностей, сопровождающихся парестезиями и повышением концентрации креатининфосфокиназы, С-реактивного белка и миоглобина, активности ACT. Все симптомы передозировки и изменения лабораторных показателей исчезают после прекращения применения препарата. Выведение препарата из организма с помощью диализа маловероятно. Однако основной гидролизный метаболит вилдаглиптина (LAY151) может быть удален из организма путем гемодиализа. Метформин Отмечено несколько случаев передозировки метформином, в том числе в результате приема внутрь препарата в количестве более 50 г. При передозировке метформином примерно в 10% случаев наблюдалась гипогликемия (однако ее связь с приемом препарата не установлена): в 32% случаев отмечался лактоацидоз. Ранними симптомами лактоацидоза являются тошнота, рвота, диарея, снижение температуры тела, боль в животе, боль в мышцах, в дальнейшем может отмечаться учащение дыхания, головокружение, нарушение сознания и развитие комы. Метформин выводится из крови с помощью гемодиализа (с клиренсом до 170 мл/мин) без развития гемодинамических нарушений. Таким образом, гемодиализ может применяться для выведения метформина из крови при передозировке препаратом. В случае передозировки необходимо проводить соответствующее симптоматическое лечение, основываясь на состоянии пациента и клинических проявлениях. Взаимодействие с другими ЛС: Вилдаглиптин и Метформин При одновременном применении вилдаглиптина (100 мг 1 раз в сутки) и метформина (1000 мг 1 раз в сутки) клинически значимых фармакокинетических взаимодействий между ними отмечено не было. Ни в ходе клинических исследований, ни в ходе широкого клинического применения препарата Галвус Мет у пациентов, одновременно получавших другие препараты и вещества, непредвиденных взаимодействий выявлено не было. Вилдаглиптин Вилдаглиптин обладает низким потенциалом лекарственного взаимодействия. Поскольку вилдаглиптин не является субстратом ферментов системы цитохрома Р450 (CYP), а также не ингибирует и не индуцирует эти изоферменты, его взаимодействие с лекарственными препаратами, которые являются субстратами, ингибиторами или индукторами Р450 (CYP), маловероятно. При одновременном применении вилдаглиптин не влияет на скорость метаболизма препаратов, являющихся субстратами ферментов: CYP1A2, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1 и CYP3A4/5. Клинического значимого взаимодействия вилдаглиптина с препаратами, наиболее часто применяемыми при лечении сахарного диабета 2 типа (глибенкламидом, пиоглитазоном, метформином) или обладающими узким терапевтическим диапазоном (амлодипином, дигоксином, рамиприлом, симвастатином, валсартаном, варфарином), не установлено. Метформин Фуросемид увеличивает Сmах и AUC метформина, но не влияет на его почечный клиренс. Метформин снижает Сmах и AUC фуросемида и также не влияет на его почечный клиренс. Нифедипин увеличивает всасывание, Сmах и AUC метформина: кроме того, он увеличивает выведение его почками. Метформин практически не влияет на фармакокинетические параметры нифедипина. Глибенкламид не влияет на фармакокинетические/фармакодинамические параметры метформина. Метформин, в целом, снижает Сmax и AUC глибенкламида, однако величина эффекта сильно варьирует. По этой причине клиническое значение подобного взаимодействия остается неясным. Органические катионы, например, амилорид, дигоксин, морфин, прокаинамид, хинидин, хинин, ранитидин, триамтерен, триметоприм, ванкомицин и др, выводящиеся почками путем канальцевой секреции, теоретически могут взаимодействовать с метформином за счет конкуренции за общие системы транспорта почечных канальцев: так, циметидин увеличивает как концентрацию метформина в плазме крови, так и его AUC на 60% и 40% соответственно. Метформин не влияет на фармакокинетические параметры циметидина. Следует соблюдать осторожность при применении препарата Галвус Мет вместе с препаратами, влияющими на функцию почек или распределение метформина в организме. Другие препараты - некоторые препараты могут вызывать гипергликемию и способствуют снижению эффективности гипогликемических средств. К подобным препаратам относятся тиазиды и другие диуретики, глюкокортикостероиды, фенотиазины, препараты гормонов щитовидной железы, эстрогены, пероральные контрацептивы, фенитоин, никотиновая кислота, симпатомиметики, антагонисты кальция и изониазид. При одновременном применении подобных препаратов или, напротив, в случае их отмены рекомендуется тщательно контролировать эффективность метформина (его гипогликемическое действие) и, в случае необходимости, корректировать дозу препарата. Не рекомендуется одновременный прием даназола во избежание гипергликемического действия последнего. При необходимости лечения даназолом и после прекращения приема последнего требуется коррекция дозы метформина под контролем концентрации глюкозы крови. Хлорпромазин при применении в больших дозах (100 мг в день) повышает гликемию, снижая высвобождение инсулина. При лечении нейролептиками и после прекращения приема последних требуется коррекция дозы препарата Галвус Мет под контролем концентрации глюкозы крови. Йодосодержащие рентгеноконтрастные средства: радиологическое исследование с применением йодсодержащих рентгеноконтрастных средств может вызывать развитие лактоацидоза у пациентов с сахарным диабетом на фоне функциональной почечной недостаточности. Применяемые в виде инъекций β-2 симпатомиметики: повышают гликемию вследствие стимуляции β-2-адренорецепторов. В этом случае необходим контроль гликемии. При необходимости рекомендуется применение инсулина. При одновременном применении метформина с производными сульфонилмочевины, инсулином, акарбозой, салицилатами возможно усиление гипогликемического действия. Поскольку при применении метформина у пациентов с острой алкогольной интоксикацией повышается риск развития лактоацидоза (в особенности, при голодании, истощении или печеночной недостаточности), при лечении препаратом Галвус Мет следует воздерживаться от употребления алкоголя и лекарственных средств, содержащих этиловый спирт. Фармакологическое действие и фармакокинетика В состав препарата Галвус Мет входят два гипогликемических средства с разными механизмами действия: вилдаглиптин, относящийся к классу ингибиторов дипептидилпептидазы-4 (ДПП-4), и метформин (в форме гидрохлорида), представитель класса бигуанидов. Комбинация этих компонентов позволяет более эффективно контролировать концентрацию глюкозы крови у пациентов с сахарным диабетом 2 типа в течение 24 часов. Вилдаглиптин, представитель класса стимуляторов островкового аппарата поджелудочной железы, селективно ингибирует фермент ДПП-4, разрушающий глюкагоноподобный пептид 1 типа (ГПП-1) и глюкозозависимый инсулинотропный полипептид (ГИП). Метформин снижает продукцию глюкозы печенью, уменьшает всасывание глюкозы в кишечнике и снижает инсулинорезистентность за счет усиления захвата и утилизации глюкозы периферическими тканями. Метформин индуцирует внутриклеточный синтез гликогена, действуя на гликогенсинтетазу, и усиливает транспорт глюкозы некоторыми мембранными белками-переносчиками глюкозы (GLUT-1 и GLUT-4). Вилдаглиптин Быстрое и полное ингибирование активности ДПП-4 после приема вилдаглиптина вызывает повышение как базальной, так и стимулированной приемом пищи секреции ГПП-1 и ГИП из кишечника в системный кровоток на протяжении всего дня. Повышая концентрацию ГПП-1 и ГИП, вилдаглиптин вызывает увеличение чувствительности β-клеток поджелудочной железы к глюкозе, что приводит к улучшению глюкозозависимой секреции инсулина. Степень улучшения функции β-клеток зависит от степени их исходного повреждения: так, у лиц без сахарного диабета (с нормальной концентрацией глюкозы в плазме крови) вилдаглиптин не стимулирует секрецию инсулина и не снижает концентрацию глюкозы. Повышая концентрацию эндогенного ГПП-1, вилдаглиптин увеличивает чувствительность α-клеток к глюкозе, что приводит к улучшению глюкозозависимой регуляции секреции глюкагона. Снижение повышенной концентрации глюкагона после еды, в свою очередь, вызывает снижение инсулинорезистентности. Увеличение соотношения инсулин/глюкагон на фоне гипергликемии, обусловленное повышением концентрации ГПП-1 и ГИП, вызывает уменьшение продукции глюкозы печеные как во время, так и после приема пищи, что приводит к снижению концентрации глюкозы в плазме крови. Кроме того, на фоне применения вилдаглиптина отмечалось снижение концентрации липидов в плазме крови после приема пищи, однако этот эффект не связан с его действием на ГПП-1 или ГИП и улучшением функции островковых клеток поджелудочной железы. Известно, что повышение концентрации ГПП-1 может приводить к замедлению опорожнения желудка, однако на фоне применения вилдаглиптина подобного эффекта не наблюдается. При применении вилдаглиптина у 5759 пациентов с сахарным диабетом 2 типа в течение 52 недель в монотерапии или в комбинации с метформином, производными сульфонилмочевины, тиазолидиндионом, или инсулином отмечалось достоверное длительное снижение концентрации гликированного гемоглобина (НbА1С) и глюкозы крови натощак. Метформин Метформин улучшает толерантность к глюкозе у пациентов с сахарным диабетом 2 типа, снижая концентрацию глюкозы в плазме как до, так и после еды. В отличие от производных сульфонилмочевины, метформин не вызывает гипогликемии ни у пациентов с сахарным диабетом 2 типа, ни у здоровых лиц (за исключением особых случаев). Терапия препаратом не приводит к развитию гиперинсулинемии. При применении метформина секреция инсулина не меняется, в то время как концентрация инсулина в плазме крови натощак и в течение дня могут снижаться. При применении метформина отмечается благоприятное влияние на метаболизм липопротеидов: снижение концентрации общего холестерина, холестерина липопротеидов низкой плотности и триглицеридов, не связанное с влиянием препарата на концентрацию глюкозы в плазме крови. Вилдаглиптин+Метформин При применении комбинированной терапии вилдаглиптином и метформином в суточных дозах 1500-3000 мг метформина и 50 мг вилдаглиптина 2 раза в сутки в течение 1 года наблюдалось статистически значимое стойкое снижение концентрации глюкозы крови (определяемое по уменьшению показателя НbА1С) и увеличение доли пациентов, у которых снижение концентрации НbА1С составило не менее 0,6-0,7% (по сравнению с группой пациентов, продолжавших получать только метформин). У пациентов, получавших комбинацию вилдаглиптина и метформина, статистически значимого изменения массы тела по сравнению с исходным состоянием не отмечалось. Через 24 недели после начала лечения в группах пациентов, получавших вилдаглиптин в комбинации с метформином, отмечалось снижение систолического и диастолического артериального давления у пациентов с артериальной гипертензией. При применении комбинации вилдаглиптина и метформина в качестве начальной терапии пациентов с сахарным диабетом 2 типа в течение 24 недель отмечалось дозозависимое снижение показателей НbА1С в сравнении с монотерапией данными препаратами. Случаи развития гипогликемии были минимальны в обеих группах терапии. При применении вилдаглиптина (50 мг 2 раза в сутки) одновременно с/без метформина в комбинации с инсулином (средняя доза 41 ВД) у пациентов в клиническом исследовании показатель НbА1С статистически значимо снижался на 0,72% (начальный показатель, в среднем 8,8%). Частота гипогликемии у пациентов, получавших лечение, была сравнима с частотой гипогликемии в группе плацебо. При применении вилдаглиптина (50 мг 2 раза в сутки) одновременно с метформином ( 1500 мг) в комбинации с глимепиридом ( 4 мг в день) у пациентов в клиническом исследовании показатель НbА1С статистически значимо снизился на 0,76% (со среднего уровня 8,8%). Фармакокинетика: Вилдаглиптин Всасывание При приеме внутрь натощак вилдаглиптин быстро всасывается, а его максимальная концентрация в плазме крови (Сmах) достигается через 1,75 часа после приема. При одновременном приеме с пищей скорость абсорбции вилдаглиптина снижается незначительно: отмечается уменьшение Сmах на 19% и увеличение времени ее достижения до 2,5 часов. Однако прием пищи не оказывает влияния на степень абсорбции и площадь под кривой "концентрация-время" (AUC). Вилдаглиптин быстро всасывается, а его абсолютная биодоступность после приема внутрь составляет 85%. Сmах и AUC в терапевтическом диапазоне доз увеличиваются примерно пропорционально дозе. Распределение Степень связывания вилдаглиптина с белками плазмы крови низкая (9,3%). Препарат распределяется равномерно между плазмой и эритроцитами. Распределение вилдаглиптина происходит предположительно экстраваскулярно, объем распределения в равновесном состоянии после внутривенного введения (Vss) составляет 71 л. Метаболизм Биотрансформация является основным путем выведения вилдаглиптина. В организме человека подвергается превращению 69% дозы препарата. Основной метаболит - LAY151 (57% дозы) - фармакологически неактивен и является продуктом гидролиза циано-компонента. Около 4% дозы препарата подвергаются амидному гидролизу. В экспериментальных исследованиях отмечается положительное влияние ДПП-4 на гидролиз препарата. Вилдаглиптин не метаболизируется при участии изоферментов системы цитохрома Р450. По данным исследований in vitro, вилдаглиптин не является субстратом изоферментов Р (СYP)450, не ингибирует и не индуцирует изоферменты системы цитохрома CYP450. Выведение После приема внутрь препарата около 85% дозы выводится почками и 15% через кишечник, почечная экскреция неизмененного вилдаглиптина составляет 23%. При внутривенном введении средний период полувыведения достигает 2 часов, общий плазменный клиренс и почечный клиренс вилдаглиптина составляют 41 л/ч и 13 л/ч соответственно. Период полувыведения (Т1/2) после приема внутрь составляет около 3 часов независимо от дозы. Фармакокинетика в особых случаях Пол, индекс массы тела и этническая принадлежность не оказывают влияния на фармакокинетику вилдаглиптина. Пациенты с нарушением функции печени У пациентов с нарушением функции печени легкой и средней степени тяжести (6-10 баллов по классификации Чайлд-Пью) после однократного применения препарата отмечается снижение биодоступности вилдаглиптина на 8% и 20% соответственно. У пациентов с нарушением функции печени тяжелой степени (12 баллов по классификации Чайлд-Пью) биодоступность вилдаглиптина повышается на 22%. Максимальное изменение биодоступности вилдаглиптина, увеличение или уменьшение в среднем до 30%, не является клинически значимым. Корреляции между степенью тяжести нарушений функции печени и биодоступностью препарата не выявлено. Пациенты с нарушением функции почек У пациентов с нарушением функции почек легкой, средней или тяжелой степени AUC вилдаглиптина увеличивалась по сравнению со здоровыми добровольцами в 1,4, 1,7 и 2 раза соответственно. AUC метаболита LAY151 увеличивалась в 1,6, 3,2, и 7,3 раз, а метаболита BQS867 - в 1,4, 2,7, и 7,3 раз у пациентов с нарушением функции почек легкой, средней и тяжелой степени соответственно. Ограниченные данные у пациентов с терминальной стадией хронической болезни почек (ХБП) указывают на то, что показатели данной группы схожи с таковыми у пациентов с нарушением функции почек тяжелой степени. Концентрация метаболита LAY151 у пациентов с терминальной стадией ХБП увеличивалась в 2-3 раза по сравнению с концентрацией у пациентов с нарушением функции почек тяжелой степени. Выведение вилдаглиптина при гемодиализе ограничено (3% при проведении процедуры длительностью более чем 3-4 часов через 4 часа после однократного приема препарата). Применение у пациентов в возрасте 65лет Максимальное увеличение биодоступности препарата на 32% (увеличение Сmах на 18%) у пациентов старше 70 лет не является клинически значимым и не влияет на ингибирование ДПП-4. Применение у пациентов в возрасте младше 18 лет Фармакокинетические особенности вилдаглиптина у детей и подростков младше 18 лет не установлены. Метформин Всасывание Абсолютная биодоступность метформина при приеме внутрь в дозе 500 мг натощак составляла 50-60%. Максимальная концентрация в плазме (Сmах) достигается через 1,81-2,69 часа после приема. При увеличении дозы препарата от 500 мг до 1500 мг, либо в дозах от 850 мг до 2250 мг внутрь, отмечалось более медленное увеличение фармакокинетических параметров (чем следовало бы ожидать для линейной зависимости). Этот эффект обусловлен не столько изменением выведения препарата, сколько замедлением его всасывания. На фоне приема пищи степень и скорость всасывания метформина также несколько снижались. Так, при однократном приеме препарата в дозе 850 мг вместе с пищей отмечалось снижение Сmах и AUC примерно на 40% и 25% и увеличение времени достижения максимальной концентрации (Тmах) на 35 минут. Клиническое значение данных фактов не установлено. Распределение При однократном приеме внутрь в дозе 850 мг кажущийся объем распределения метформина составляет 654±358 л. Препарат практически не связывается с белками плазмы, в то время как производные сульфонилмочевины связываются с ними более чем на 90%. Метформин проникает в эритроциты (вероятно усиление этого процесса со временем). При применении метформина по стандартной схеме (стандартные доза и частота приема) равновесная концентрация препарата в плазме крови достигается в течение 24-48 часов и, как правило, не превышает 1 мкг/мл. В ходе контролируемых клинических исследований Сmах метформина в плазме крови не превышала 5 мкг/мл (даже при приеме в высоких дозах). Метаболизм При однократном внутривенном введении метформина здоровым добровольцам, он выводится почками в неизмененном виде. При этом препарат не метаболизируется в печени (у человека не выявлено никаких метаболитов) и не выводится с желчью. Выведение Поскольку почечный клиренс метформина примерно в 3,5 раза превышает клиренс креатинина (КК), основным путем выведения препарата является канальцевая секреция. При приеме внутрь примерно 90% всосавшейся дозы выводится почками в течен
1743 Руб.Тонометр автомат Омрон M2 Классик НЕМ-7117-RU (на предплечье)

Тонометр Omron M2 Classic, модель HEM 7117-RU, - это полностью автоматизированный измеритель артериального давления, работающий на основе осциллометрического метода. Прибор легко и быстро измеряет артериальное давление и частоту пульса, используя технологию Intelli. Sense, которая обеспечивает комфортное для пациента управляемое нагнетание воздуха в манжету без предварительной установки требуемого уровня давления воздуха или его повторной накачки. Осенью 2016 года в продажу поступил тонометр Omron M2 Classic, модель HEM 7122-LRU, нового поколения, который пришел на смену тонометру Omron M2 Classic, модель HEM 7117-RU. Тонометр Omron M2 Classic, модель HEM 7122-LRU, обладает расширенным по сравнению с моделью предыдущего поколения Omron M2 Classic набором потребительских функций: обновленной системой интеллектуального управления прибором Intellisense, индикатором правильной фиксации манжеты, расширенным объемом памяти на 60 последних измерений, а также предусмотренной возможностью подключения детской манжеты Omron CS2 (17 - 22 см). В комплект поставки тонометра Omron M2 Classic, модель HEM 7122-LRU, входит универсальная компрессионная манжета Omron Easy Cuff (22 - 42 см.), нового поколения. Отличительные особенности: Технология управления Intellisense - интеллектуальная адаптация тонометра под каждого пользователя Крупный трехстрочный жидкокристаллический дисплей Объем памяти рассчитан на сохранение 30 результатов измерения Интегрированный индикатор повышенного давления Индикатор аритмии Измерение выполняется еще в процессе накачивания манжеты воздухом Компрессионная манжета Omron CM, повторяющая форму руки, средний размер, 22 - 32 см. В комплекте чехол для хранения и переноски Прошел клиническую апробацию, нет возрастных ограничений к применению данного тонометра Работает от батареек, набор которых идет в комплекте Технические характеристики: Наименование прибора: цифровой автоматический измеритель артериального давления Модель: Omron M2 Classic (HEM 7117-RU) Технология интеллектуального измерения: Intellisense Способ измерения: осциллометрический Экран: высококонтрастный матричный жидкокристаллический дисплей Диапазон измерений давления: 0-299 мм рт.ст, частоты пульса: 40-180 ударов/мин. Предельная погрешность измерения давление: не более 3 мм рт.ст, частота пульса: не более 5% Клинически апробирован: Да, IP Манжета в комплекте: Компрессионная манжета Omron CM для рук с длиной окружности плеча от 22 до 32 см. Возможность использования большой манжеты (32-42 см): Есть, манжета Omron CL Возможность использование детской манжеты (17-22 см): Нет Индикатор аритмии: Есть Индикатор движения: Нет Индикатор повышенного давления: Есть Графический индикатор уровня АД: Нет Индикатор правильной фиксации манжеты: Нет Датчик правильного положения руки: Нет Индикатор утренней гипертензии: Нет Индикатор двойной проверки точности: Нет Звуковой сигнал (Отключаемая функция): Нет Расчет среднего значения последних измерений: Нет Запись даты/времени измерения: Нет Подсветка экрана: Нет Подключения к компьютеру: Нет Память для 2-х пользователей: Нет Сетевой адаптер в комплекте: Нет Вес электронного блока (без элементов питания): 290 гр Размеры электронного блока: 104 X 84 X 129 мм Память: 30 измерений Нагнетание воздуха: автоматическое с помощью воздушного электрического компрессора Выпуск воздуха: автоматический Условия эксплуатации: температура воздуха от +10˚C до +40˚C при относительной влажности от 30% до 85% Условия хранения: температура воздуха от -20˚C до +60˚C при относительной влажности от 10% до 95% Питание: 4 элемента AAA (мизинчиковые батарейки) Срок службы элементов питания: 300 измерений с частотой два измерения в день Срок службы электронного блока: 10 лет Срок службы манжеты: 1 год Гарантия: 5 лет Комплектация: Электронный блок тонометра Omron M2 Classic (HEM 7117-RU) Компрессионная манжета Omron CM (22 - 32 см.) Руководство по эксплуатации Чехол для хранения прибора Комплект элементов питания Гарантийный талон Журнал для записи артериального давления
2544 Руб.Галвус Мет таб. п/о 50мг+1000мг №30
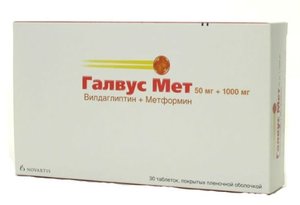
Показания Сахарный диабет 2 типа (в сочетании с диетотерапией и физическими упражнениями): - при недостаточной эффективности монотерапии вилдаглиптином или метформином: - у пациентов, ранее получающих комбинированную терапию вилдаглиптином и метформином в виде монопрепаратов: - в комбинации с производными сульфонилмочевины (тройная комбинированная терапия) у пациентов, ранее получавших терапию производными сульфонилмочевины и метформином без достижения адекватного контроля гликемии: - в тройной комбинированной терапии с инсулином у пациентов, ранее получавших инсулинотерапию в стабильной дозе и метформин без достижения адекватного контроля гликемии: - в качестве начальной терапии у пациентов с сахарным диабетом 2 типа при недостаточной эффективности диетотерапии, физических упражнений и при необходимости улучшения контроля гликемии. Противопоказания Противопоказания - Повышенная чувствительность к вилдаглиптину или метформину или любым другим компонентам препарата: - почечная недостаточность или нарушения функции почек (при концентрации креатинина в сыворотке крови 1,5 мг% (>:135 мкмол/л) для мужчин и 1,4 мг% (>:110 мкмол/л) для женщин): - острые состояния, с риском развития нарушения функции почек: дегидратация (при диарее, рвоте), лихорадка, тяжелые инфекционные заболевания, состояния гипоксии (шок, сепсис, инфекции почек, бронхолегочные заболевания): - острая и хроническая сердечная недостаточность, острый инфаркт миокарда, острая сердечно-сосудистая недостаточность (шок), дыхательная недостаточность: - нарушения функции печени: - острый или хронический метаболический ацидоз (включая диабетический кетоацидоз в сочетании с комой или без таковой). Диабетический кетоацидоз должен корректироваться инсулинотерапией. Лактоацидоз (в том числе и в анамнезе): - препарат не следует применять за 48 часов перед хирургическими операциями, радиоизотопными, рентгенологическими исследованиями с введением контрастных средств и в течение 48 часов после их проведения: - беременность и период грудного вскармливания: - сахарный диабет 1 типа: - хронический алкоголизм, острое отравление алкоголем: - соблюдение низкокалорийной диеты (менее 1000 ккал/сут): Эффективность и безопасность применения препарата у детей до 18 лет не установлена. Поскольку у пациентов с нарушением функции печени в ряде случаев отмечался лактоацидоз, возможно, являющийся одним из побочных эффектов метформина, препарат Галвус Мет не следует применять у пациентов с заболеваниями печени или нарушениями биохимических показателей функции печени. С осторожностью: Препараты, содержащие метформин, рекомендуется применять с осторожностью у пациентов старше 60 лет при выполнении тяжелой физической работы, в связи с повышенным риском развития у них лактоацидоза. Беременность Беременность В экспериментальных исследованиях у животных при применении вилдаглиптина в дозах, в 200 раз превышающих рекомендуемые, препарат не вызывал нарушения раннего развития эмбриона и не оказывал тератогенного действия. При применении вилдаглиптина в комбинации с метформином в соотношении 1:10 также не было выявлено тератогенного действия. Поскольку достаточных данных по применению препарата Галвус Мет при беременности нет, применение препарата при беременности противопоказано. Грудное вскармливание Метформин проникает в грудное молоко. Неизвестно, выделяется ли вилдаглиптин с грудным молоком. Применение препарата Галвус Мет в период грудного вскармливания противопоказано. Применение и дозы Препарат применяют внутрь. Режим дозирования препарата Галвус Мет следует подбирать индивидуально в зависимости от эффективности и переносимости терапии. При применении препарата Галвус Мет не следует превышать рекомендованную максимальную суточную дозу вилдаглиптина (100 мг). Рекомендуемую начальную дозу препарата Галвус Мет следует подбирать, учитывая длительность течения сахарного диабета и уровень гликемии, состояние пациента и уже применявшиеся у пациента схемы лечения вилдаглиптином и/или метформином. Для уменьшения выраженности побочных эффектов со стороны органов желудочно-кишечного тракта, характерных для метформина, препарат Галвус Мет принимают во время еды. Начальная доза препарата Галвус Мет при неэффективности монотерапии вилдаглиптином Лечение препаратом Галвус Мет можно начинать с одной таблетки дозировкой 50 мг+500 мг 2 раза в сутки: после оценки терапевтического эффекта дозу можно постепенно увеличивать. Начальная доза препарата Галвус Мет при неэффективности монотерапии метформином В зависимости от дозы уже принимаемого метформина, лечение препаратом Галвус Мет можно начинать с одной таблетки дозировкой 50 мг+500 мг, 50 мг+850 мг или 50 мг+1000 мг 2 раза в сутки. Начальная доза препарата Галвус Мет у пациентов, ранее получавших комбинированную терапию вилдаглиптином и метформином в виде отдельных таблеток В зависимости от доз уже принимаемых вилдаглиптина или метформина, лечение препаратом Галвус Мет следует начинать с таблетки, максимально близкой по дозировке к существующему лечению, 50 мг+500 мг, 50 мг+850 мг или 50 мг+1000 мг, и корректировать дозу в зависимости от эффективности. Стартовая доза препарата Галвус Мет в качестве начальной терапии у пациентов с сахарным диабетом 2 типа при недостаточной эффективности диетотерапии и физических упражнений В качестве стартовой терапии препарат Галвус Мет следует применять в начальной дозе 50 мг+500 мг однократно в сутки и после оценки терапевтического эффекта постепенно увеличивать дозу до 50 мг+1000 мг дважды в сутки. Комбинированная терапия препаратом Галвус Мет и производными сульфонилмочевины или инсулином Доза препарата Галвус Мет рассчитывается, исходя из дозы вилдаглиптина 50 мг х 2 раза в день (100 мг в день) и метформина в дозе, равной принимаемой ранее в виде монопрепарата. Пациенты с нарушением функции почек У пациентов с нарушением функции почек может потребоваться коррекция дозы препарата при клиренсе креатинина (КК, рассчитанном по формуле Кокрофта-Голта) в диапазоне от 60 до 90 мл/мин. Применение препарата Галвус Мет у пациентов с КК <:60 мл/мин противопоказано. Применение у пациентов в возрасте 65 лет Метформин выводится почками. Поскольку у пациентов старше 65 лет часто отмечается нарушение функции почек, дозу препарата Галвус Мет у данных пациентов следует корректировать, основываясь на показателях функции почек. При применении препарата у пациентов старше 65 лет необходимо регулярно контролировать функцию почек. Применение у пациентов в возрасте младше 18 лет Поскольку безопасность и эффективность препарата Галвус Мет у детей и подростков младше 18 лет не изучена, применение препарата противопоказано у данной категории пациентов. Побочные эффекты и передозировка Побочные эффекты: Представленные ниже данные относятся к применению вилдаглиптина и метформина в монотерапии и в комбинации. На фоне терапии вилдаглиптином редко отмечались нарушения функции печени (включая гепатит) бессимптомного течения. В большинстве случаев данные нарушения и отклонения показателей функции печени от нормы разрешались самостоятельно без осложнений после прекращения терапии препаратом. При применении вилдаглиптина в дозе 50 мг 1 или 2 раза в сутки частота повышения активности "печеночных" ферментов (аланинаминотрансферазы (АЛТ) или аспартатаминотрансферазы (ACT) в 3 раза выше верхней границы нормы (ВГН)) составляла 0,2% или 0,3% соответственно (по сравнению с 0,2% в контрольной группе). Повышение активности "печеночных" ферментов в большинстве случаев было бессимптомным, не прогрессировало и не сопровождалось холестазом или желтухой. Для оценки частоты развития нежелательных явлений (НЯ) использовались следующие критерии: очень часто ( 1/10), часто ( 1/100, <:1/10), нечасто ( 1/1000, <:1/100), редко ( 1/10,000,<:1/1,000), очень редко (<:1/10,000), включая отдельные случаи. Нежелательные реакции, возможно связанные с применением комбинированной терапии вилдаглиптином и метформином (частота развития которых в группе вилдаглиптина и метформина отличалась от таковой на фоне применения плацебо и метформина более чем на 2%) представлены ниже. Нарушения со стороны нервной системы: часто - головная боль, головокружение, тремор. При применении вилдаглиптина в комбинации с метформином в различных дозах гипогликемия отмечалась в 0,9% случаев (для сравнения в группе плацебо в комбинации с метформином - в 0,4%). Частота НЯ со стороны желудочно- кишечного тракта на фоне комбинированной терапии вилдаглиптином и метформином составляла 12,9%. При применении метформина подобные НЯ отмечались у 18,1% пациентов.В группах пациентов, получавших метформин в комбинации с вилдаглиптином, нарушения со стороны желудочно-кишечного тракта отмечались с частотой 10-15%, а в группе пациентов, получавших метформин в комбинации с плацебо, - с частотой 18%. Долгосрочные клинические исследования продолжительностью до 2 лет не выявили каких-либо дополнительных отклонений профиля безопасности или непредвиденных рисков при применении вилдаглиптина в монотерапии. Изучение применения комбинации вилдаглиптина и метформина в качестве стартовой терапии при сахарном диабете 2 типа не выявило рисков и дополнительных данных по безопасности применения. При применении вилдаглиптина одновременно с инсулином В контролируемых клинических исследованиях при применении вилдаглиптина в дозе 50 мг 2 раза в день в комбинации с инсулином в сочетании с метформином или без него, частота отмены терапии в связи с развитием побочных реакций составила 0,3% в группе вилдаглиптина, при этом в группе плацебо случаев отмены терапии не было. Частота гипогликемии была сопоставимой в обеих группах (14,0% в группе вилдаглиптина и 16,4% в группе плацебо).В группе вилдаглиптина отмечены случаи гипогликемии тяжелой степени у двух пациентов, в группе плацебо - у 6 пациентов. На момент завершения исследования вилдаглиптин не оказывал влияния на среднюю массу тела (масса тела увеличена на +0,6 кг по сравнению с исходной в группе вилдаглиптина, в группе плацебо изменений не отмечено).НЯ у пациентов, получавших вилдаглиптин 50 мг 2 раза в день в комбинации с инсулином (с метформином или без него), представлены ниже. Нарушения со стороны нервной системы: часто - головная боль. Нарушения со стороны желудочно-кишечного тракта: часто - тошнота, гастроэзофагеальный рефлюкс: нечасто - диарея, метеоризм. Нарушения со стороны обмена веществ и питания: часто - гипогликемия. Общие расстройства и нарушения в месте введения: часто - озноб. При применении вилдаглиптина в комбинации с препаратами сульфонилмочевины. Случаев отмены препарата, связанных с развитием НЯ в группе комбинированной терапии вилдаглиптином, метформином и глимепиридом, отмечено не было. В группе комбинированной терапии плацебо, метформином и глимепиридом частота НЯ составила 0,6%. Гипогликемия отмечалась часто в обеих группах (5,1% в группе комбинированной терапии вилдаглиптином, метформином и глимепиридом и 1,9% в группе комбинированной терапии плацебо, метформином и глимепиридом). В группе вилдаглиптина отмечен один эпизод гипогликемии тяжелой степени. На момент завершения исследования значимого влияния на массу тела выявлено не было (+0,6 кг в группе вилдаглиптина и -0,1 кг в группе плацебо). НЯ у пациентов, получавших вилдаглиптин 50 мг 1 или 2 раза в день в комбинации препаратами сульфонилмочевины, представлены ниже (с метформином или без него). Инфекционные и паразитарные заболевания: очень редко - назофарингит. Нарушения со стороны нервной системы: часто - головокружение, тремор, астения. Нарушения со стороны желудочно-кишечного тракта: нечасто - запор. Нарушения со стороны обмена веществ и питания: часто - гипогликемия. Нарушения со стороны кожи и подкожных тканей: часто - гипергидроз. При применении вилдаглиптина в монотерапии. Инфекционные и паразитарные заболевания: очень редко - инфекции верхних дыхательных путей, назофарингит. Нарушения со стороны нервной системы: часто - головокружение: нечасто головная боль. Нарушения со стороны желудочно-кишечного тракта: нечасто - запор. Нарушения со стороны кожи и подкожных тканей: нечасто - кожная сыпь. Нарушения со стороны скелетно-мышечной и соединительной ткани: часто - артралгия. Нарушения со стороны сосудов: нечасто - периферические отеки. При применении комбинированной терапии вилдаглиптином и метформином не отмечалось клинически значимого повышения частоты вышеуказанных НЯ, отмечавшихся при приеме вилдаглиптина. На фоне монотерапии вилдаглиптином или метформином частота развития гипогликемии составляла 0,4% (нечасто). Монотерапия вилдаглиптином и комбинированное лечение вилдаглиптин+метформин не оказывали влияния на массу тела пациентов. Долгосрочные клинические исследования продолжительностью до 2 лет не выявили каких-либо дополнительных отклонений профиля безопасности или непредвиденных рисков при применении вилдаглиптина в монотерапии. Пострегистрационные исследования В пострегистрационном периоде выявлены следующие побочные реакции (поскольку данные сообщаются в добровольном порядке от популяции неопределенного размера, достоверно определить частоту развития данных НЯ не представляется возможным, в связи с чем они классифицированы как частота неизвестна): гепатит (обратим при прекращении терапии), повышение активности "печеночных" ферментов, миалгия, крапивница, панкреатит, буллезное и эксфолиативное поражения кожи, артралгия, в редких случаях выраженная. При применении метформина в монотерапии. Нарушения со стороны обмена веществ и питания: очень часто - снижение аппетита: очень редко - лактоацидоз. Нарушения со стороны желудочно-кишечного тракта: очень часто - метеоризм, тошнота, рвота, диарея, боль в животе: часто - дисгевзия. Нарушения со стороны печени и желчевыводящих путей: очень редко - гепатит. Нарушения со стороны кожи и подкожных тканей: очень редко - кожные реакции (в частности, эритема, зуд, крапивница). Лабораторные и инструментальные данные: очень редко - уменьшение всасывания витамина В12, изменение показателей функции печени. Уменьшение всасывания витамина В12 и снижение его концентрации в сыворотке крови на фоне применения метформина отмечалось очень редко у пациентов, получавших препарат в течение длительного времени, и, как правило, не представляло клинического значения. Следует учитывать возможность уменьшения всасывания витамина В12 у пациентов с мегалобластной анемией. Отдельные случаи гепатита, которые наблюдались на фоне применения метформина, разрешались после его отмены. Если отмечено ухудшение типического течения любого из указанных в инструкции побочных эффектов или Вы заметили любые другие побочные эффекты, не указанные в инструкции, сообщите об этом врачу. Передозировка: Вилдаглиптин Вилдаглиптин хорошо переносится в дозе до 200 мг/сут. При применении препарата в дозе 400 мг/сут могут наблюдаться боли в мышцах, редко - легкие транзиторные парестезии, лихорадка, отеки и транзиторное повышение активности липазы (выше ВГН в 2 раза). При увеличении лозы вилдаглиптина до 600 мг/сут возможно развитие отеков конечностей, сопровождающихся парестезиями и повышением концентрации креатининфосфокиназы, С-реактивного белка и миоглобина, активности ACT. Все симптомы передозировки и изменения лабораторных показателей исчезают после прекращения применения препарата. Выведение препарата из организма с помощью диализа маловероятно. Однако основной гидролизный метаболит вилдаглиптина (LAY151) может быть удален из организма путем гемодиализа. Метформин Отмечено несколько случаев передозировки метформином, в том числе в результате приема внутрь препарата в количестве более 50 г. При передозировке метформином примерно в 10% случаев наблюдалась гипогликемия (однако ее связь с приемом препарата не установлена): в 32% случаев отмечался лактоацидоз. Ранними симптомами лактоацидоза являются тошнота, рвота, диарея, снижение температуры тела, боль в животе, боль в мышцах, в дальнейшем может отмечаться учащение дыхания, головокружение, нарушение сознания и развитие комы. Метформин выводится из крови с помощью гемодиализа (с клиренсом до 170 мл/мин) без развития гемодинамических нарушений. Таким образом, гемодиализ может применяться для выведения метформина из крови при передозировке препаратом. В случае передозировки необходимо проводить соответствующее симптоматическое лечение, основываясь на состоянии пациента и клинических проявлениях. Взаимодействие с другими ЛС: Вилдаглиптин и Метформин При одновременном применении вилдаглиптина (100 мг 1 раз в сутки) и метформина (1000 мг 1 раз в сутки) клинически значимых фармакокинетических взаимодействий между ними отмечено не было. Ни в ходе клинических исследований, ни в ходе широкого клинического применения препарата Галвус Мет у пациентов, одновременно получавших другие препараты и вещества, непредвиденных взаимодействий выявлено не было. Вилдаглиптин Вилдаглиптин обладает низким потенциалом лекарственного взаимодействия. Поскольку вилдаглиптин не является субстратом ферментов системы цитохрома Р450 (CYP), а также не ингибирует и не индуцирует эти изоферменты, его взаимодействие с лекарственными препаратами, которые являются субстратами, ингибиторами или индукторами Р450 (CYP), маловероятно. При одновременном применении вилдаглиптин не влияет на скорость метаболизма препаратов, являющихся субстратами ферментов: CYP1A2, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1 и CYP3A4/5. Клинического значимого взаимодействия вилдаглиптина с препаратами, наиболее часто применяемыми при лечении сахарного диабета 2 типа (глибенкламидом, пиоглитазоном, метформином) или обладающими узким терапевтическим диапазоном (амлодипином, дигоксином, рамиприлом, симвастатином, валсартаном, варфарином), не установлено. Метформин Фуросемид увеличивает Сmах и AUC метформина, но не влияет на его почечный клиренс. Метформин снижает Сmах и AUC фуросемида и также не влияет на его почечный клиренс. Нифедипин увеличивает всасывание, Сmах и AUC метформина: кроме того, он увеличивает выведение его почками. Метформин практически не влияет на фармакокинетические параметры нифедипина. Глибенкламид не влияет на фармакокинетические/фармакодинамические параметры метформина. Метформин, в целом, снижает Сmax и AUC глибенкламида, однако величина эффекта сильно варьирует. По этой причине клиническое значение подобного взаимодействия остается неясным. Органические катионы, например, амилорид, дигоксин, морфин, прокаинамид, хинидин, хинин, ранитидин, триамтерен, триметоприм, ванкомицин и др, выводящиеся почками путем канальцевой секреции, теоретически могут взаимодействовать с метформином за счет конкуренции за общие системы транспорта почечных канальцев: так, циметидин увеличивает как концентрацию метформина в плазме крови, так и его AUC на 60% и 40% соответственно. Метформин не влияет на фармакокинетические параметры циметидина. Следует соблюдать осторожность при применении препарата Галвус Мет вместе с препаратами, влияющими на функцию почек или распределение метформина в организме. Другие препараты - некоторые препараты могут вызывать гипергликемию и способствуют снижению эффективности гипогликемических средств. К подобным препаратам относятся тиазиды и другие диуретики, глюкокортикостероиды, фенотиазины, препараты гормонов щитовидной железы, эстрогены, пероральные контрацептивы, фенитоин, никотиновая кислота, симпатомиметики, антагонисты кальция и изониазид. При одновременном применении подобных препаратов или, напротив, в случае их отмены рекомендуется тщательно контролировать эффективность метформина (его гипогликемическое действие) и, в случае необходимости, корректировать дозу препарата. Не рекомендуется одновременный прием даназола во избежание гипергликемического действия последнего. При необходимости лечения даназолом и после прекращения приема последнего требуется коррекция дозы метформина под контролем концентрации глюкозы крови. Хлорпромазин при применении в больших дозах (100 мг в день) повышает гликемию, снижая высвобождение инсулина. При лечении нейролептиками и после прекращения приема последних требуется коррекция дозы препарата Галвус Мет под контролем концентрации глюкозы крови. Йодосодержащие рентгеноконтрастные средства: радиологическое исследование с применением йодсодержащих рентгеноконтрастных средств может вызывать развитие лактоацидоза у пациентов с сахарным диабетом на фоне функциональной почечной недостаточности. Применяемые в виде инъекций β-2 симпатомиметики: повышают гликемию вследствие стимуляции β-2-адренорецепторов. В этом случае необходим контроль гликемии. При необходимости рекомендуется применение инсулина. При одновременном применении метформина с производными сульфонилмочевины, инсулином, акарбозой, салицилатами возможно усиление гипогликемического действия. Поскольку при применении метформина у пациентов с острой алкогольной интоксикацией повышается риск развития лактоацидоза (в особенности, при голодании, истощении или печеночной недостаточности), при лечении препаратом Галвус Мет следует воздерживаться от употребления алкоголя и лекарственных средств, содержащих этиловый спирт. Фармакологическое действие и фармакокинетика В состав препарата Галвус Мет входят два гипогликемических средства с разными механизмами действия: вилдаглиптин, относящийся к классу ингибиторов дипептидилпептидазы-4 (ДПП-4), и метформин (в форме гидрохлорида), представитель класса бигуанидов. Комбинация этих компонентов позволяет более эффективно контролировать концентрацию глюкозы крови у пациентов с сахарным диабетом 2 типа в течение 24 часов. Вилдаглиптин, представитель класса стимуляторов островкового аппарата поджелудочной железы, селективно ингибирует фермент ДПП-4, разрушающий глюкагоноподобный пептид 1 типа (ГПП-1) и глюкозозависимый инсулинотропный полипептид (ГИП). Метформин снижает продукцию глюкозы печенью, уменьшает всасывание глюкозы в кишечнике и снижает инсулинорезистентность за счет усиления захвата и утилизации глюкозы периферическими тканями. Метформин индуцирует внутриклеточный синтез гликогена, действуя на гликогенсинтетазу, и усиливает транспорт глюкозы некоторыми мембранными белками-переносчиками глюкозы (GLUT-1 и GLUT-4). Вилдаглиптин Быстрое и полное ингибирование активности ДПП-4 после приема вилдаглиптина вызывает повышение как базальной, так и стимулированной приемом пищи секреции ГПП-1 и ГИП из кишечника в системный кровоток на протяжении всего дня. Повышая концентрацию ГПП-1 и ГИП, вилдаглиптин вызывает увеличение чувствительности β-клеток поджелудочной железы к глюкозе, что приводит к улучшению глюкозозависимой секреции инсулина. Степень улучшения функции β-клеток зависит от степени их исходного повреждения: так, у лиц без сахарного диабета (с нормальной концентрацией глюкозы в плазме крови) вилдаглиптин не стимулирует секрецию инсулина и не снижает концентрацию глюкозы. Повышая концентрацию эндогенного ГПП-1, вилдаглиптин увеличивает чувствительность α-клеток к глюкозе, что приводит к улучшению глюкозозависимой регуляции секреции глюкагона. Снижение повышенной концентрации глюкагона после еды, в свою очередь, вызывает снижение инсулинорезистентности. Увеличение соотношения инсулин/глюкагон на фоне гипергликемии, обусловленное повышением концентрации ГПП-1 и ГИП, вызывает уменьшение продукции глюкозы печеные как во время, так и после приема пищи, что приводит к снижению концентрации глюкозы в плазме крови. Кроме того, на фоне применения вилдаглиптина отмечалось снижение концентрации липидов в плазме крови после приема пищи, однако этот эффект не связан с его действием на ГПП-1 или ГИП и улучшением функции островковых клеток поджелудочной железы. Известно, что повышение концентрации ГПП-1 может приводить к замедлению опорожнения желудка, однако на фоне применения вилдаглиптина подобного эффекта не наблюдается. При применении вилдаглиптина у 5759 пациентов с сахарным диабетом 2 типа в течение 52 недель в монотерапии или в комбинации с метформином, производными сульфонилмочевины, тиазолидиндионом, или инсулином отмечалось достоверное длительное снижение концентрации гликированного гемоглобина (НbА1С) и глюкозы крови натощак. Метформин Метформин улучшает толерантность к глюкозе у пациентов с сахарным диабетом 2 типа, снижая концентрацию глюкозы в плазме как до, так и после еды. В отличие от производных сульфонилмочевины, метформин не вызывает гипогликемии ни у пациентов с сахарным диабетом 2 типа, ни у здоровых лиц (за исключением особых случаев). Терапия препаратом не приводит к развитию гиперинсулинемии. При применении метформина секреция инсулина не меняется, в то время как концентрация инсулина в плазме крови натощак и в течение дня могут снижаться. При применении метформина отмечается благоприятное влияние на метаболизм липопротеидов: снижение концентрации общего холестерина, холестерина липопротеидов низкой плотности и триглицеридов, не связанное с влиянием препарата на концентрацию глюкозы в плазме крови. Вилдаглиптин+Метформин При применении комбинированной терапии вилдаглиптином и метформином в суточных дозах 1500-3000 мг метформина и 50 мг вилдаглиптина 2 раза в сутки в течение 1 года наблюдалось статистически значимое стойкое снижение концентрации глюкозы крови (определяемое по уменьшению показателя НbА1С) и увеличение доли пациентов, у которых снижение концентрации НbА1С составило не менее 0,6-0,7% (по сравнению с группой пациентов, продолжавших получать только метформин). У пациентов, получавших комбинацию вилдаглиптина и метформина, статистически значимого изменения массы тела по сравнению с исходным состоянием не отмечалось. Через 24 недели после начала лечения в группах пациентов, получавших вилдаглиптин в комбинации с метформином, отмечалось снижение систолического и диастолического артериального давления у пациентов с артериальной гипертензией. При применении комбинации вилдаглиптина и метформина в качестве начальной терапии пациентов с сахарным диабетом 2 типа в течение 24 недель отмечалось дозозависимое снижение показателей НbА1С в сравнении с монотерапией данными препаратами. Случаи развития гипогликемии были минимальны в обеих группах терапии. При применении вилдаглиптина (50 мг 2 раза в сутки) одновременно с/без метформина в комбинации с инсулином (средняя доза 41 ВД) у пациентов в клиническом исследовании показатель НbА1С статистически значимо снижался на 0,72% (начальный показатель, в среднем 8,8%). Частота гипогликемии у пациентов, получавших лечение, была сравнима с частотой гипогликемии в группе плацебо. При применении вилдаглиптина (50 мг 2 раза в сутки) одновременно с метформином ( 1500 мг) в комбинации с глимепиридом ( 4 мг в день) у пациентов в клиническом исследовании показатель НbА1С статистически значимо снизился на 0,76% (со среднего уровня 8,8%). Фармакокинетика: Вилдаглиптин Всасывание При приеме внутрь натощак вилдаглиптин быстро всасывается, а его максимальная концентрация в плазме крови (Сmах) достигается через 1,75 часа после приема. При одновременном приеме с пищей скорость абсорбции вилдаглиптина снижается незначительно: отмечается уменьшение Сmах на 19% и увеличение времени ее достижения до 2,5 часов. Однако прием пищи не оказывает влияния на степень абсорбции и площадь под кривой "концентрация-время" (AUC). Вилдаглиптин быстро всасывается, а его абсолютная биодоступность после приема внутрь составляет 85%. Сmах и AUC в терапевтическом диапазоне доз увеличиваются примерно пропорционально дозе. Распределение Степень связывания вилдаглиптина с белками плазмы крови низкая (9,3%). Препарат распределяется равномерно между плазмой и эритроцитами. Распределение вилдаглиптина происходит предположительно экстраваскулярно, объем распределения в равновесном состоянии после внутривенного введения (Vss) составляет 71 л. Метаболизм Биотрансформация является основным путем выведения вилдаглиптина. В организме человека подвергается превращению 69% дозы препарата. Основной метаболит - LAY151 (57% дозы) - фармакологически неактивен и является продуктом гидролиза циано-компонента. Около 4% дозы препарата подвергаются амидному гидролизу. В экспериментальных исследованиях отмечается положительное влияние ДПП-4 на гидролиз препарата. Вилдаглиптин не метаболизируется при участии изоферментов системы цитохрома Р450. По данным исследований in vitro, вилдаглиптин не является субстратом изоферментов Р (СYP)450, не ингибирует и не индуцирует изоферменты системы цитохрома CYP450. Выведение После приема внутрь препарата около 85% дозы выводится почками и 15% через кишечник, почечная экскреция неизмененного вилдаглиптина составляет 23%. При внутривенном введении средний период полувыведения достигает 2 часов, общий плазменный клиренс и почечный клиренс вилдаглиптина составляют 41 л/ч и 13 л/ч соответственно. Период полувыведения (Т1/2) после приема внутрь составляет около 3 часов независимо от дозы. Фармакокинетика в особых случаях Пол, индекс массы тела и этническая принадлежность не оказывают влияния на фармакокинетику вилдаглиптина. Пациенты с нарушением функции печени У пациентов с нарушением функции печени легкой и средней степени тяжести (6-10 баллов по классификации Чайлд-Пью) после однократного применения препарата отмечается снижение биодоступности вилдаглиптина на 8% и 20% соответственно. У пациентов с нарушением функции печени тяжелой степени (12 баллов по классификации Чайлд-Пью) биодоступность вилдаглиптина повышается на 22%. Максимальное изменение биодоступности вилдаглиптина, увеличение или уменьшение в среднем до 30%, не является клинически значимым. Корреляции между степенью тяжести нарушений функции печени и биодоступностью препарата не выявлено. Пациенты с нарушением функции почек У пациентов с нарушением функции почек легкой, средней или тяжелой степени AUC вилдаглиптина увеличивалась по сравнению со здоровыми добровольцами в 1,4, 1,7 и 2 раза соответственно. AUC метаболита LAY151 увеличивалась в 1,6, 3,2, и 7,3 раз, а метаболита BQS867 - в 1,4, 2,7, и 7,3 раз у пациентов с нарушением функции почек легкой, средней и тяжелой степени соответственно. Ограниченные данные у пациентов с терминальной стадией хронической болезни почек (ХБП) указывают на то, что показатели данной группы схожи с таковыми у пациентов с нарушением функции почек тяжелой степени. Концентрация метаболита LAY151 у пациентов с терминальной стадией ХБП увеличивалась в 2-3 раза по сравнению с концентрацией у пациентов с нарушением функции почек тяжелой степени. Выведение вилдаглиптина при гемодиализе ограничено (3% при проведении процедуры длительностью более чем 3-4 часов через 4 часа после однократного приема препарата). Применение у пациентов в возрасте 65лет Максимальное увеличение биодоступности препарата на 32% (увеличение Сmах на 18%) у пациентов старше 70 лет не является клинически значимым и не влияет на ингибирование ДПП-4. Применение у пациентов в возрасте младше 18 лет Фармакокинетические особенности вилдаглиптина у детей и подростков младше 18 лет не установлены. Метформин Всасывание Абсолютная биодоступность метформина при приеме внутрь в дозе 500 мг натощак составляла 50-60%. Максимальная концентрация в плазме (Сmах) достигается через 1,81-2,69 часа после приема. При увеличении дозы препарата от 500 мг до 1500 мг, либо в дозах от 850 мг до 2250 мг внутрь, отмечалось более медленное увеличение фармакокинетических параметров (чем следовало бы ожидать для линейной зависимости). Этот эффект обусловлен не столько изменением выведения препарата, сколько замедлением его всасывания. На фоне приема пищи степень и скорость всасывания метформина также несколько снижались. Так, при однократном приеме препарата в дозе 850 мг вместе с пищей отмечалось снижение Сmах и AUC примерно на 40% и 25% и увеличение времени достижения максимальной концентрации (Тmах) на 35 минут. Клиническое значение данных фактов не установлено. Распределение При однократном приеме внутрь в дозе 850 мг кажущийся объем распределения метформина составляет 654±358 л. Препарат практически не связывается с белками плазмы, в то время как производные сульфонилмочевины связываются с ними более чем на 90%. Метформин проникает в эритроциты (вероятно усиление этого процесса со временем). При применении метформина по стандартной схеме (стандартные доза и частота приема) равновесная концентрация препарата в плазме крови достигается в течение 24-48 часов и, как правило, не превышает 1 мкг/мл. В ходе контролируемых клинических исследований Сmах метформина в плазме крови не превышала 5 мкг/мл (даже при приеме в высоких дозах). Метаболизм При однократном внутривенном введении метформина здоровым добровольцам, он выводится почками в неизмененном виде. При этом препарат не метаболизируется в печени (у человека не выявлено никаких метаболитов) и не выводится с желчью. Выведение Поскольку почечный клиренс метформина примерно в 3,5 раза превышает клиренс креатинина (КК), основным путем выведения препарата является канальцевая секреция. При приеме внутрь примерно 90% всосавшейся дозы выводится почками в течен
2129 Руб.Липопрайм таб. п/о 20мг №30

Показания Первичная гиперхолестеринемия по классификации Фредриксона (тип IIа, включая семейную гетерозиготную гиперхолестеринемию) или смешанная гиперхолестеринемия (тип IIb) в качестве дополнения к диете, когда диета и другие немедикаментозные методы лечения (например, физические упражнения, снижение массы тела) оказываются недостаточными. - Семейная гомозиготная гиперхолестеринемия в качестве дополнения к диете и другой липидснижающей терапии (например, ЛПНП-аферез), или в случаях, когда подобная терапия недостаточно эффективна. - Гипертриглицеридемия (тип IV по классификации Фредриксона) в качестве дополнения к диете. - Для замедления прогрессирования атеросклероза в качестве дополнения к диете у пациентов, которым показана терапия для снижения концентрации общего ХС и ХС-ЛПНП. - Первичная профилактика основных сердечно-сосудистых осложнений (инсульта, инфаркта, артериальной реваскуляризации) у взрослых пациентов без клинических признаков ишемической болезни сердца (ИБС), но с повышенным риском ее развития (возраст старше 50 лет для мужчин и старше 60 лет для женщин, повышенная концентрация С-реактивного белка ( 2 мг/л) при наличии, как минимум одного из дополнительных факторов риска, таких как артериальная гипертензия, низкая концентрация ХС-ЛПВП, курение, семейный анамнез раннего начала ИБС). Противопоказания Противопоказания Для применения в суточной дозе 5 мг, 10 мг и 20 мг: - повышенная чувствительность к розувастатину или любому из компонентов препарата: - заболевания печени в активной фазе, включая стойкое повышение сывороточной активности трансаминаз и любое повышение активности трансаминаз в сыворотке крови (более чем в 3 раза по сравнению с верхней границей нормы): - тяжелые нарушения функции почек (КК менее 30 мл/мин): - миопатия: - одновременный приём циклоспорина: - у женщин: беременность, период грудного вскармливания, отсутствие адекватных методов контрацепции: - пациентам, предрасположенным к развитию миотоксических осложнений: - непереносимость лактозы, дефицит лактазы или глюкозо-галактозная мальабсорбция (препарат содержит лактозу). Для применения в суточной дозе 40 мг: - повышенная чувствительность к розувастатину или любому из компонентов препарата: - одновременный приём циклоспорина: - заболевания печени в активной фазе, включая стойкое повышение сывороточной активности трансаминаз и любое повышение активности трансаминаз в сыворотке крови (более чем в 3 раза по сравнению с верхней границей нормы): - пациенты с печеночной недостаточностью: - пациентам с факторами риска развития миопатии/рабдомиолиза, а именно: почечная недостаточность умеренной степени тяжести (КК менее 60 мл/мин): гипотиреоз: личный или семейный анамнез мышечных заболеваний: миотоксичность на фоне приёма других ингибиторов ГМГ-КоА-редуктазы или фибратов в анамнезе: - у женщин: беременность, период грудного вскармливания, отсутствие адекватных методов контрацепции: - чрезмерное употребление алкоголя: - состояния, которые могут приводить к повышению плазменной концентрации розувастатина: - одновременный приём фибратов: - пациентам азиатской расы: - непереносимость лактозы, дефицит лактазы или глюкозо-галактозная мальабсорбция (препарат содержит лактозу). С осторожностью: Для применения в суточной дозе 5 мг, 10 мг и 20 мг: Наличие риска развития миопатии/рабдомиолиза - почечная недостаточность, гипотиреоз, личный или семейный анамнез наследственных мышечных заболеваний и предшествующий анамнез мышечной токсичности при применении других ингибиторов ГМГ-КоА-редуктазы или фибратов: чрезмерное употребление алкоголя: возраст старше 65 лет: состояния, при которых отмечено повышение плазменной концентрации розувастатина: расовая принадлежность (азиатская раса): одновременное назначение с фибратами (см. раздел "Фармакокинетика"): заболевания печени в анамнезе: сепсис: артериальная гипотензия: обширные хирургические вмешательства, травмы, тяжелые метаболические, эндокринные или водно-электролитные нарушения или неконтролируемые судорожные припадки. Для применения в суточной дозе 40 мг: Почечная недостаточность легкой степени тяжести (КК более 60 мл/мин): возраст старше 65 лет: заболевания печени в анамнезе: сепсис: артериальная гипотензия: обширные хирургические вмешательства, травмы, тяжелые метаболические, эндокринные или водно-электролитные нарушения или неконтролируемые судорожные припадки. Применение в педиатрической практике Эффективность и безопасность применения препарата у детей до 18 лет не установлена. Опыт применения препарата в педиатрической практике ограничен небольшим количеством детей (от 8 лет и старше) с семейной гомозиготной гиперхолестеринемией. В настоящее время не рекомендуется применять препарат Липопрайм у детей до 18 лет. Пациенты с печеночной недостаточностью. Данные или опыт применения препарата у пациентов с баллом выше 9 по шкале Чайлд-Пью отсутствует. Беременность Липопрайм противопоказан при беременности и в период грудного вскармливания. Женщины репродуктивного возраста, принимающие Липопрайм, должны применять адекватные методы контрацепции. Поскольку холестерин и другие продукты биосинтеза холестерина важны для развития плода, потенциальный риск ингибирования ГМГ-КоА редуктазы превышает пользу от применения препарата у беременных. В случае диагностирования беременности в процессе терапии прием препарата должен быть прекращен немедленно. Данные в отношении выделения розувастатина с грудным молоком отсутствуют, поэтому в период грудного кормления прием препарата необходимо прекратить. Применение и дозы Внутрь, не разжевывать, проглатывать целиком, запивая водой. Препарат может назначаться в любое время суток независимо от времени приема пищи. До начала терапии препаратом Липопрайм пациент должен начать соблюдать стандартную гипохолестеринемическую диету и продолжать соблюдать ее во время лечения. Доза препарата должна подбираться индивидуально в зависимости от целей терапии и терапевтического ответа на лечение, принимая во внимание текущие рекомендации по целевой концентрации липидов. Рекомендуемая начальная доза для пациентов, начинающих принимать препарат, или для пациентов, переведенных с приема других ингибиторов ГМГ-КоА редуктазы, должна составлять 5 или 10 мг препарата Липопрайм 1 раз в сутки. При выборе начальной дозы следует руководствоваться индивидуальной концентрацией холестерина и принимать во внимание возможный риск сердечно-сосудистых осложнений, а также необходимо оценивать потенциальный риск развития побочных эффектов. В случае необходимости, доза может быть увеличена до большей через 4 недели. В связи с возможным развитием побочных эффектов при приёме дозы 40 мг по сравнению с более низкими дозами препарата, увеличение дозы до 40 мг (см. раздел "Побочное действие") после дополнительного приема дозы выше рекомендуемой начальной дозы в течение 4-х недель терапии, может проводиться только у пациентов с тяжелой степенью гиперхолестеринемии и с высоким риском сердечно-сосудистых осложнений (особенно у пациентов с семейной гиперхолестеринемией), у которых не был достигнут желаемый результат терапии при приёме дозы 20 мг, и которые будут находиться под наблюдением специалиста. Рекомендуется особенно тщательное наблюдение за пациентами, получающими препарат в дозе 40 мг. Не рекомендуется назначение дозы 40 мг пациентам, ранее не обращавшимся к врачу. После 2-4-х недель терапии и/или при повышении дозы препарата Липопрайм необходим контроль показателей липидного обмена (при необходимости требуется коррекция дозы). Пожилые пациенты Не требуется коррекции дозы. Пациенты с почечной недостаточностью У пациентов с почечной недостаточностью легкой или умеренной степени тяжести коррекции дозы не требуется. Противопоказано применение всех дозировок препарата Липопрайм у пациентов с тяжелой почечной недостаточностью (КК менее 30 мл/мин.). Противопоказано применение препарата в дозе 40 мг пациентами с умеренными нарушениями функции почек (КК менее 30 мл/мин). Пациентам с умеренными нарушениями функции почек рекомендуется начальная доза препарата 5 мг. Пациенты с печеночной недостаточностью Липопрайм противопоказан пациентам с заболеваниями печени в активной фазе (см. раздел "Противопоказания"). Особые популяции. Этнические группы При изучении фармакокинетических параметров розувастатина у пациентов, принадлежащих к разным этническим группам, отмечено увеличение системной концентрации розувастатина среди японцев и китайцев. Следует учитывать данный факт при назначении препарата Липопрайм данным группам пациентов. При назначении доз 10 и 20 мг рекомендуемая начальная доза для пациентов азиатской расы составляет 5 мг. Противопоказано назначение препарата в дозе 40 мг пациентам монголоидной расы (см. раздел "Противопоказания"). Пациенты, предрасположенные к миопатии. Противопоказано назначение препарата в дозе 40 мг пациентам с факторами, которые могут указывать на предрасположенность к развитию миопатии (см. раздел "Противопоказания"). При назначении доз 10 и 20 мг рекомендуемая начальная доза для данной группы пациентов составляет 5 мг (см. раздел "Противопоказания"). Генетический полиморфизм У носителей генотипов SLC01B1 (ОАТР1В1) С.521СС и ABCG2 (BCRP) С.421АА отмечалось увеличение экспозиции (AUC) к розувастатину в 1,6 и 2,4 раза, соответственно, по сравнению с носителями генов SLC01B1 с.52ПТ и ABCG2 С.421СС. Для пациентов-носителей генотипов С.521СС или С.421АА рекомендуемая максимальная доза препарата Липопрайм составляет 20 мг 1 раз в сутки (см. разделы "Фармакокинетика", "Особые указания" и "Взаимодействие с другими лекарственными средствами"). Сопутствующая терапия Розувастатин связывается с различными транспортными белками (в частности, с ОАТР1В1 и BCRP). При одновременном применении препарата Липопрайм с лекарственными препаратами (такими как циклоспорин, некоторые ингибиторы протеазы ВИЧ, включая комбинацию ритонавира с атазанавиром, лопинавиром и/или типранавиром), повышающими концентрацию розувастатина в плазме крови за счет взаимодействия с транспортными белками, может повышаться риск миопатии (включая рабдомиолиз). Следует ознакомиться с инструкцией по применению этих препаратов перед их назначением одновременно с препаратом Липопрайм. В таких случаях следует оценить возможность назначения альтернативной терапии или временного прекращения приема препарата Липопрайм. Если же применение указанных выше препаратов необходимо, следует оценить соотношение пользы и риска сопутствующей терапии препаратом Липопрайм и рассмотреть возможность снижения его дозы (см. раздел "Взаимодействие с другими лекарственными средствами"). Побочные эффекты и передозировка Побочные эффекты: Побочные эффекты, наблюдаемые при приеме розувастатина, обычно выражены незначительно и проходят самостоятельно. Как и при применении других ингибиторов ГМГ-КоА-редуктазы, частота возникновения побочных эффектов носит дозозависимый характер. Частота возникновения нежелательных эффектов представлена следующим образом, согласно классификации Всемирной организации здравоохранения: часто ( 1/100, <: 1/10): нечасто ( 1/1000, <: 1/100): редко ( 1/10 000, <: 1/1 000): очень редко (<: 1/10 000), включая отдельные сообщения: неуточненная частота (не может быть подсчитана по имеющимся данным). Иммунная система Редко: реакции повышенной чувствительности, включая ангионевротический отек. Со стороны эндокринной системы Часто: сахарный диабет 2 типа. Со стороны центральной нервной системы Часто: головная боль, головокружение. Со стороны пищеварительного тракта Часто: запор, тошнота, боли в животе. Редко: панкреатит. Со стороны кожных покровов Нечасто: кожный зуд, сыпь, крапивница. Со стороны опорно-двигательного аппарата Часто: миалгия. Редко: миопатия (включая миозит), рабдомиолиз. Прочие Часто: астенический синдром. Со стороны мочевыводящей системы У пациентов может выявляться протеинурия. Изменения количества белка в моче (от отсутствия или следовых количеств до + или больше) наблюдаются у менее 1% пациентов, получающих 10 - 20 мг препарата, и у приблизительно 3% пациентов, получающих 40 мг препарата. Незначительное изменение количества белка в моче отмечалось при приеме дозы 20 мг. В большинстве случаев протеинурия уменьшается или исчезает в процессе терапии и не означает возникновения острого или прогрессирования существующего заболевания почек. Со стороны опорно-двигательного аппарата При применении розувастатина во всех дозировках и, в особенности при приёме доз препарата, превышающих 20 мг, сообщалось о следующих воздействиях на опорно-двигательный аппарат: миалгия, миопатия (включая миозит), в редких случаях рабдомиолиз с острой почечной недостаточностью или без нее. Дозозависимое повышение активности креатинфосфокиназы (КФК) наблюдается у незначительного числа пациентов, принимавших розувастатин. В большинстве случаев оно было незначительным, бессимптомным и временным. В случае повышения активности КФК (более чем в 5 раз по сравнению с верхней границей нормы (ВГН)) терапия должна быть приостановлена (см. раздел "Особые указания"). Со стороны печени При применении розувастатина наблюдается дозозависимое повышение активности "печеночных" трансаминаз у незначительного числа пациентов. В большинстве случаев оно незначительно, бессимптомно и временно. Лабораторные показатели: Повышение концентрации глюкозы, билирубина, активности гамма- глутамилтранспептидазы, щелочной фосфатазы, нарушения функции щитовидной железы. Постмаркетинговое применение Сообщалось о следующих побочных эффектах в постмаркетинговом применении препарата: Со стороны системы кроветворения Неуточненной частоты: тромбоцитопения. Со стороны пищеварительного тракта Очень редко: желтуха, гепатит. Редко: повышение активности "печеночных" трансаминаз. Неуточненной частоты: диарея. Со стороны опорно-двигательного аппарата Очень редко: артралгия. Неуточненной частоты: иммуноопосредованная некротизирующая миопатия. Со стороны центральной нервной системы Очень редко: потеря или снижение памяти. Неуточненной частоты: периферическая нейропатия. Со стороны дыхательной системы Неуточненной частоты: кашель, одышка. Со стороны мочевыводящей системы Очень редко: гематурия. Со стороны кожных покровов и подкожно-жировой клетчатки Неуточненной частоты: синдром Стивенса-Джонсона. Со стороны репродуктивной системы Очень редко: гинекомастия. Прочие Неуточненной частоты: периферические отеки. При применении некоторых статинов сообщалось о следующих побочных эффектах: депрессия, нарушения сна, включая бессонницу и "кошмарные" сновидения, сексуальная дисфункция, гипергликемия, повышение концентрации гликозилированного гемоглобина. Сообщалось о единичных случаях интерстициального заболевания легких, особенно при длительном применении препаратов. Передозировка: Симптомы: при одновременном приеме нескольких суточных доз фармакокинетические параметры розувастатина не изменяются. Лечение: специфического лечения при передозировке розувастатином не существует. При передозировке рекомендуется проводить симптоматическое лечение и мероприятия, направленные на поддержание функций жизненно важных органов и систем. Необходим контроль функции печени и активности КФК. Маловероятно, что гемодиализ будет эффективен. Взаимодействие с другими ЛС: Ингибиторы транспортных белков: розувастатин связывается с некоторыми транспортными белками, в частности, с ОАТР1В1 и BCRP. Сопутствующее применение препаратов, которые являются ингибиторами транспортных белков, может сопровождаться увеличением концентрации розувастатина в плазме крови и повышенным риском развития миопатии (см. таблицу 1 и разделы "Способ применения и дозы" и "Особые указания"). Циклоспорин: при одновременном применении розувастатина и циклоспорина AUC розувастатина была в среднем в 7 раз выше значения, которое отмечалось у здоровых добровольцев (см. таблицу 1). Не влияет на плазменную концентрацию циклоспорина. Липопрайм противопоказан пациентам, принимающим циклоспорин (см. раздел "Противопоказания"). Гемфиброзил и другие гиполипидемические средства: совместное применение розувастатина и гемфиброзила приводит к увеличению в 2 раза максимальной концентрации розувастатина в плазме крови и AUC розувастатина. Основываясь на данных по специфическому взаимодействию, не ожидается фармакокинетически значимого взаимодействия с фенофибратом, возможно фармакодинамическое взаимодействие. Гемфиброзил, фенофибрат, другие фибраты и липидснижающие дозы никотиновой кислоты (более 1 г/сутки) увеличивали риск возникновения миопатии при одновременном применении с ингибиторами ГМГ-КоА-редуктазы, возможно в связи с тем, что они могут вызывать миопатию и при применении в монотерапии (см. раздел "Особые указания"). При одновременном приёме препарата с гемфиброзилом, фибратами, никотиновой кислотой в липидснижающих дозах (более 1 г/сутки) пациентам рекомендуется начальная доза препарата 5 мг. Прием в дозе 40 мг противопоказан при одновременном применении с фибратами (см. разделы "Противопоказания", "Способ применения и дозы", "Особые указания"). Эзетимиб: одновременное применение розувастатина в дозе 10 мг и эзетимиба в дозе 10 мг не сопровождалось изменением AUC розувастатина у пациентов с гиперхолестеринемией (см. таблицу 1). Нельзя исключить увеличение риска возникновения побочных эффектов из-за фармакодинамического взаимодействия между розувастатином и эзетимибом. Ингибиторы протеазы вируса иммунодефицита человека (ВИЧ): несмотря на то, что точный механизм взаимодействия неизвестен, совместный приём ингибиторов протеазы ВИЧ может приводить к значительному увеличению экспозиции розувастатина (см. таблицу 1). Фармакокинетическое исследование по одновременному применению 20 мг розувастатина с комбинированным препаратом, содержащим два ингибитора протеазы ВИЧ (400 мг лопинавира/100 мг ритонавира) у здоровых добровольцев приводило к приблизительно двукратному и пятикратному увеличению AUC (о-24) и Сmах розувастатина, соответственно. Поэтому одновременный прием розувастатина и ингибиторов протеазы ВИЧ не рекомендуется (см. разделы "Способ применения и дозы" и "Особые указания", таблицу 1). Антациды: одновременное применение розувастатина и суспензий антацидов, содержащих алюминий и магния гидроксид, приводит к снижению плазменной концентрации розувастатина примерно на 50%. Данный эффект выражен слабее, если антациды применяются через 2 часа после приема розувастатина. Клиническое значение подобного взаимодействия не изучалось. Эритромицин: одновременное применение розувастатина и эритромицина приводит к уменьшению AUC розувастатина на 20 % и Сmах розувастатина на 30%. Подобное взаимодействие может возникать в результате усиления моторики кишечника, вызываемого приёмом эритромицина. Изоферменты цитохрома Р450: результаты исследований in vivo и in vitro показали, что розувастатин не является ни ингибитором, ни индуктором изоферментов цитохрома Р450. Кроме того, розувастатин является слабым субстратом для этих изоферментов. Не было отмечено клинически значимого взаимодействия между розувастатином и флуконазолом (ингибитором изоферментов CYP2C9 и CYP3A4) и кетоконазолом (ингибитором изоферментов CYP2A6 CYP3A4). Таким образом, не ожидается взаимодействия розувастатина с другими лекарственными средствами на уровне метаболизма с участием изоферментов цитохрома Р450. Фузидовая кислота: исследований по изучению взаимодействия розувастатина и фузидовой кислоты не проводилось. Как и при приеме других статинов, были получены постмаркетинговые сообщения о случаях рабдомиолиза при одновременном приеме розувастатина и фузидовой кислоты. Необходимо наблюдение пациентов. При необходимости, возможно временное прекращение приема розувастатина. Взаимодействие с лекарственными средствами, которое требует коррекции дозы розувастатина (см. таблицу 1). Дозу препарата Липопрайм следует корректировать при необходимости его одновременного применения с лекарственными средствами, увеличивающими экспозицию к розувастатину. Следует ознакомиться с инструкцией по применению этих препаратов перед их назначением одновременно с препаратом Липопрайм. Если ожидается увеличение экспозиции в 2 раза и более, начальная доза препарата Липопрайм должна составлять 5 мг 1 раз в сутки. Также следует корректировать максимальную суточную дозу препарата Липопрайм так, чтобы ожидаемая экспозиция к розувастатину, не превышала таковую для дозы 40 мг, принимаемой без одновременного назначения лекарственных средств, взаимодействующих с розувастатином. Например, максимальная суточная доза препарата Липопрайм при одновременном применении с гемфиброзилом составляет 20 мг (увеличение экспозиции в 1,9 раза), с ритонавиром/атазанавиром - 10 мг (увеличение экспозиции в 3,1 раза). Таблица 1 Влияние сопутствующей терапии на экспозицию к розувастатину (AUC, данные приведены в порядке убывания) ТАБЛИЦЪ Влияние применения розувастатина на другие препараты Антагонисты витамина К: начало терапии розувастатином или увеличение дозы препарата у пациентов, получающих одновременно антагонисты витамина К (например, варфарин), может приводить к увеличению Международного Нормализованного Отношения (МНО). Отмена розувастатина или снижение дозы а и препарата может приводить к уменьшению МНО. В таких случаях рекомендуется контроль МНО. Пероральные контрацептивы/гормонозаместительная терапия: одновременное применение розувастатина и пероральных контрацептивов увеличивает AUC этинилэстрадиола и AUC норгестрела на 26% и 34%, соответственно. Такое увеличение плазменной концентрации должно учитываться при подборе дозы пероральных контрацептивов. Фармакокинетические данные по одновременному применению препарата Липопрайм и гормонозаместительной терапии отсутствуют, следовательно, нельзя исключить аналогичного эффекта и при применении данного сочетания. Однако подобная комбинация широко применялась во время проведения клинических исследований и хорошо переносилась пациентами. Другие лекарственные средства: не ожидается клинически значимого взаимодействия розувастатина с дигоксином. Фармакологическое действие и фармакокинетика Механизм действия Розувастатин является селективным, конкурентным ингибитором ГМГ-КоА-редуктазы, фермента, превращающего 3-гидрокси-3-метилглутарил коэнзим А в мевалонат, предшественник холестерина. Основной мишенью действия розувастатина является печень, где осуществляется синтез холестерина (ХС) и катаболизм липопротеинов низкой плотности (ЛПНП). Розувастатин увеличивает число ‘‘печеночных” рецепторов ЛПНП на поверхности клеток, повышая захват и катаболизм ЛПНП, что в свою очередь приводит к ингибированию синтеза липопротеинов очень низкой плотности (ЛПОНП), уменьшая тем самым общее количество ЛПНП и ЛПОНП. Фармакодинамика Розувастатин снижает повышенную концентрацию холестерина-ЛПНП (ХС-ЛПНП), общего холестерина, триглицеридов (ТГ), повышает концентрацию холестерина липопротеинов высокой плотности (ХС-ЛПВП), а также снижает концентрацию аполипопротеина В (АпоВ), ХС-неЛПВП, ХС-ЛПОНП, ТГ-ЛПОНП и увеличивает уровень аполипопротеина A-I (АпоА-I), снижает соотношение ХС-ЛПНП/ХС-ЛПВП, общий ХС/ХС-ЛПВП и ХС-неЛПВП/ ХС-ЛПВП и соотношение АпоВ/АпоА-1. Терапевтический эффект появляется в течение одной недели после начала терапии розувастатином, через 2 недели лечения достигает 90% от максимально возможного эффекта. Максимальный терапевтический эффект обычно достигается к 4-ой неделе и поддерживается при регулярном приёме. Клиническая эффективность Розувастатин эффективен у взрослых пациентов с гиперхолестеринемией с или без гипертриглицеридемии: вне зависимости от расовой принадлежности, пола или возраста, в т.ч. у пациентов с сахарным диабетом и семейной гиперхолестеринемией. У 80% пациентов с гиперхолестеринемией IIа и IIb типа по классификации Фредриксона (средняя исходная концентрация ХС-ЛПНП около 4,8 ммоль/л) на фоне приема препарата в дозе 10 мг концентрация ХС-ЛПНП достигает значений менее 3 ммоль/л. У пациентов с гетерозиготной семейной гиперхолестеринемией, получающих розувастатин в дозе 20-80 мг, отмечается положительная динамика показателей липидного профиля. После титрования до суточной дозы 40 мг (12 недель терапии), отмечается снижение концентрации ХС-ЛПНП на 53%. У пациентов с гомозиготной семейной гиперхолестеринемией, принимающих розувастатин в дозе 20 мг и 40 мг, среднее снижение концентрации ХС-ЛПНП составляет 22%. Аддитивный эффект отмечается в комбинации с фенофибратом в отношении концентрации триглицеридов и с никотиновой кислотой в липидснижающих дозах (более 1 г/сутки) в отношении концентрации ХС-ЛПВП. Фармакокинетика: Абсорбция и распределение Максимальная концентрация розувастатина в плазме крови достигается приблизительно через 5 часов после приема внутрь. Абсолютная биодоступность составляет примерно 20%. Розувастатин метаболизируется преимущественно печенью, которая является основным местом синтеза холестерина и метаболизма ХС-ЛПНП. Объем распределения розувастатина составляет примерно 134 л. Приблизительно 90% розувастатина связывается с белками плазмы крови, в основном с альбумином. Метаболизм Подвергается ограниченному метаболизму (около 10%). Розувастатин является непрофильным субстратом для метаболизма изоферментами системы цитохрома Р450. Основным изоферментом, участвующим в метаболизме розувастатина, является изофермент CYP2C9. Изоферменты CYP2C19, CYP3A4 и CYP2D6 вовлечены в метаболизм в меньшей степени. Основными выявленными метаболитами розувастатина являются N-десметил и лактоновые метаболиты. N-десметилрозувастатин примерно на 50% менее активен, чем розувастатин, лактоновые метаболиты фармакологически не активны. Более 90% фармакологической активности по ингибированию циркулирующей ГМГ-КоА редуктазы обеспечивается розувастатином, остальное - его метаболитами. Выведение Около 90% дозы розувастатина выводится в неизмененном виде через кишечник (включая абсорбированный и не абсорбированный розувастатин). Оставшаяся часть выводится почками. Плазменный период полувыведения (Т1/2) составляет примерно 19 часов. Т1/2 не изменяется при увеличении дозы препарата. Средний геометрический плазменный клиренс составляет приблизительно 50 л/час (коэффициент вариации 21,7%). Как и в случае других ингибиторов ГМГ-КоА редуктазы, в процесс "печеночного" захвата розувастатина вовлечен мембранный переносчик холестерина, выполняющий важную роль в печеночной элиминации розувастатина. Линейность Системная экспозиция розувастатина увеличивается пропорционально дозе. Фармакокинетические параметры не изменяются при ежедневном приеме. Особые популяции пациентов Возраст и пол Пол и возраст не оказывают клинически значимого влияния на фармакокинетику розувастатина. Этнические группы Фармакокинетические исследования показали приблизительно двукратное увеличение медианы AUC (площади под кривой "концентрация-время") и Сmах (максимальной концентрации в плазме крови) розувастатина у пациентов азиатской национальности (японцев, китайцев, филиппинцев, вьетнамцев и корейцев) по сравнению с европейцами: у индийских пациентов показано увеличение медианы AUC и Сmах в 1,3 раза. Фармакокинетический анализ не выявил клинически значимых различий в фармакокинетике среди европейцев и представителей негроидной расы. Почечная недостаточность У пациентов с легкой и умеренно выраженной почечной недостаточностью величина плазменной концентрации розувастатина или N-десметилрозувастатина существенно не меняется. У пациентов с тяжелой почечной недостаточностью (клиренс креатинина (КК) менее 30 мл/мин) концентрация розувастатина в плазме крови в 3 раза выше, а концентрация N-десметилрозувастатина в 9 раз выше, чем у здоровых добровольцев. Концентрация розувастатина в плазме крови у пациентов на гемодиализе была примерно на 50% выше, чем у здоровых добровольцев. Печеночная недостаточность У пациентов с различной степенью печеночной недостаточности не выявлено увеличение Т1/2 розувастатина. У пациентов с баллом 7 и ниже по шкале Чайлд-Пью не выявлено увеличение Т1/2 розувастатина. У двух пациентов с баллами 8 и 9 по шкале Чайлд-Пью отмечено увеличение Т1/2 по крайней мере, в 2 раза. Опыт применения розувастатина у пациентов с баллом выше 9 по шкале Чайлд-Пью отсутствует. Генетический полиморфизм Ингибиторы ГМГ-КоА-редуктазы, в том числе и розувастатин, связываются с транспортными белками ОАТР1В1 (полипептид транспорта органических анионов, участвующих в захвате статинов гепатоцитами) BCRP (эффлюксный транспортер). У носителей генотипов SLC01B1 (ОАТР1В1) С.521СС и ABCG2 (BCRP) С.421АА отмечалось увеличение экспозиции (AUC) к розувастатину в 1,6 и 2,4 раза, соответственно, по сравнению с носителями генов SLC01B1 с.52ITT и ABCG2 С.421СС. Особые указания Почечные эффекты со стороны мочевыделительной системы У пациентов, получавших высокие дозы препарата розувастатин (в основном 40 мг), наблюдалась канальцевая протеинурия, которая в большинстве случаев была транзиторной. Такая протеинурия не свидетельствовала об остром заболевании почек или прогрессировании заболевания почек. У пациентов, принимающих препарат в дозе 40 мг, рекомендуется контролировать показатели функции почек во время лечения. Со стороны опорно-двигательного аппарата При применении препарата розувастатин во всех дозировках и, в особенности при приёме доз препарата, превышающих 20 мг, сообщалось о следующих воздействиях на опорно-двигательный аппарат: миалгия, миопатия, в редких случаях рабдомиолиз. Определение активности креатинфосфокиназы Определение КФК не следует проводить после интенсивных физических нагрузок или при наличии других возможных причин увеличения активности КФК, что может привести к неверной интерпретации полученных результатов. В случае если исходная активность КФК существенно повышен (в 5 раз выше, чем верхняя граница нормы), через 5-7 дней следует провести повторное измерение. Не следует начинать терапию, если повторный тест подтверждает исходную активность КФК (выше более чем в 5 раз по сравнению с ВГН). До начала терапии При назначении препарата, также как и при назначении других ингибиторов ГМГ-КоА-редуктазы, следует проявлять осторожность пациентам с имеющимися факторами риска миопатии/рабдомиолиза (см. раздел “С осторожностью”), необходимо рассмотреть соотношение риска и возможной пользы терапии и проводить клиническое наблюдение. Во время терапии Следует проинформировать пациента о необходимости немедленного сообщения врачу о случаях неожиданного появления мышечных болей, мышечной слабости или спазмах, особенно в сочетании с недомоганием и лихорадкой. У таких пациентов следует определять активность КФК. Терапия должна быть прекращена, если активность КФК значительно увеличен (более чем в 5 раз по сравнению с ВГН) или если симптомы со стороны мышц резко выражены и вызывают ежедневный дискомфорт (даже, если активность КФК в 5 раз меньше по сравнению с ВГН). Если симптомы исчезают, и активность КФК возвращается к норме, следует рассмотреть вопрос о повторном назначении препарата Липопрайм или других ингибиторов ГМГ-КоА-редуктазы в меньших дозах при тщательном наблюдении за пациентом. Рутинный контроль активности КФК при отсутствии симптомов нецелесообразен. Отмечены очень редкие случаи иммуноопосредованной некротизирующей миопатии с клиническими проявлениями в виде стойкой слабости проксимальных мышц и повышения активности КФК в сыворотке во время лечения или при прекращении приема статинов, в т.ч. розувастатина. Может потребоваться проведение дополнительных исследований мышечной системы и нервной системы, серологических исследований, а также терапия иммунодепрессивными средствами. Не отмечено признаков увеличения воздействия на скелетную мускулатуру при приеме препарата розувастатин и сопутствующей терапии. Однако сообщалось об увеличении числа случаев миозита и миопатии у пациентов, принимавших другие ингибиторы ГМГ-КоА-редуктазы в сочетании с производными фибриновой кислоты, включая гемфиброзил, циклоспорин, никотиновую кислоту в липидснижающих дозах (более 1 г/сутки), азольные противогрибковые средства, ингибиторы протеазы ВИЧ и макролидные антибиотики. Гемфиброзил увеличивает риск возникновения миопатии при сочетанном назначении с некоторыми ингибиторами ГМГ-КоА-редуктазы. Таким образом, не рекомендуется одновременное назначение препарата розувастатин и гемфиброзила. Должно быть тщательно взвешено соотношение риска и возможной пользы при совместном применении препарата Липопрайм и фибратов или липидснижающих доз никотиновой кислоты (более 1 г/сутки). Противопоказан приём препарата Липопрайм в дозе 40 мг совместно с фибратами. Через 2-4 недели после начала лечения и/или при повышении дозы препарата Липопрайм необходим контроль показателей липидного обмена (при необходимости требуется коррекция дозы). Со стороны печени Рекомендуется проводить определение показателей функции печени до начала терапии и через 3 месяца после начала терапии. Прием препарата Липопрайм следует прекратить или уменьшить дозу препарата, если активность "п
664 Руб.Розувастатин таб. п/о 20мг №30

Показания Первичная гиперхолестеринемия по классификации Фредриксона (тип IIа, включая семейную гетерозиготную гиперхолестеринемию) или смешанная гиперхолестеринемия (тип IIb) в качестве дополнения к диете, когда диета и другие немедикаментозные методы лечения (например, физические упражнения, снижение массы тела) оказываются недостаточными. - Семейная гомозиготная гиперхолестеринемия в качестве дополнения к диете и другой липидснижающей терапии (например, ЛПНП-аферез), или в случаях, когда подобная терапия недостаточно эффективна. - Гипертриглицеридемия (тип IV по классификации Фредриксона) в качестве дополнения к диете. - Для замедления прогрессирования атеросклероза в качестве дополнения к диете у пациентов, которым показана терапия для снижения концентрации общего ХС и ХС-ЛПНП. - Первичная профилактика основных сердечно-сосудистых осложнении (инсульта, инфаркта, артериальной реваскуляризации) у взрослых пациентов без клинических признаков ишемической болезни сердца (ИБС), но с повышенным риском ее развития (возраст старше 50 лет для мужчин и старше 60 лет для женщин, повышенная концентрация С-реактивного белка ( 2 мг/л) при наличии, как минимум одного из дополнительных факторов риска, таких как артериальная гипертензия, низкая концентрация ХС-ЛПВП, курение, семейный анамнез раннего начала ИБС). Противопоказания Противопоказания Для применения в суточной дозе 5 мг, 10 мг и 20 мг: - повышенная чувствительность к розувастатину или любому из компонентов препарата: - заболевания печени в активной фазе, включая стойкое повышение сывороточной активности трансаминаз и любое повышение активности трансаминаз в сыворотке крови (более чем в 3 раза по сравнению с верхней границей нормы): - пациенты с печеночной недостаточностью: - тяжелые нарушения функции почек (КК менее 30 мл/мин.): - миопатия: - одновременный приём циклоспорина: - детский и подростковый возраст до 18 лет: - у женщин: беременность, период грудного вскармливания, отсутствие адекватных методов контрацепции: - пациентам, предрасположенным к развитию миотоксических осложнений: - непереносимость лактозы, дефицит лактазы или глюкозо-галактозная мальабсорбция (препарат содержит лактозу). Для применения в суточной дозе 40 мг: - повышенная чувствительность к розувастатину или любому из компонентов препарата: - одновременный приём циклоспорина: - заболевания печени в активной фазе, включая стойкое повышение сывороточной активности трансаминаз и любое повышение активности трансаминаз в сыворотке крови (более чем в 3 раза по сравнению с верхней границей нормы): - пациенты с печеночной недостаточностью: - пациентам с факторами риска развития миопатии/рабдомиолиза - почечная недостаточность средней степени тяжести (КК менее 60 мл/мин): - гипотиреоз: - личный или семейный анамнез мышечных заболеваний: - миотоксичность на фоне приёма других ингибиторов ГМГ-КоА-редуктазы или фибратов в анамнезе: - у женщин: беременность, период грудного вскармливания, отсутствие адекватных методов контрацепции: - чрезмерное употребление алкоголя: - состояния, которые могут приводить к повышению плазменной концентрации розувастатина: - одновременный приём фибратов: - пациентам азиатской расы: - непереносимость лактозы, дефицит лактазы или глюкозо-галактозная мальабсорбция (препарат содержит лактозу). С осторожностью: Для применения в суточной дозе 5 мг, 10 мг и 20 мг: Наличие риска развития миопатии/рабдомиолиза - почечная недостаточность, гипотиреоз, личный или семейный анамнез наследственных мышечных заболеваний и предшествующий анамнез мышечной токсичности при применении других ингибиторов ГМГ-КоА-редуктазы или фибратов: чрезмерное употребление алкоголя: возраст старше 65 лет: состояния, при которых отмечено повышение плазменной концентрации розувастатина: расовая принадлежность (монголоидная раса): одновременное назначение с фибратами: заболевания печени в анамнезе: сепсис: артериальная гипотензия: обширные хирургические вмешательства, травмы, тяжелые метаболические, эндокринные или электролитные нарушения или неконтролируемые судорожные припадки. Для применения в суточной дозе 40 мг: Почечная недостаточность легкой степени тяжести (КК более 60 мл/мин): возраст старше 65 лет: заболевания печени в анамнезе: сепсис: артериальная гипотензия: обширные хирургические вмешательства, травмы, тяжелые метаболические, эндокринные или электролитные нарушения или неконтролируемые судорожные припадки. Применение в педиатрической практике Эффективность и безопасность применения препарата у детей до 18 лет не установлена. Опыт применения препарата в педиатрической практике ограничен небольшим количеством детей (от 8 лет и старше) семейной гомозиготной гиперхолестеринемией. В настоящее время не рекомендуется применять Розувастатин у детей до 18 лет. Беременность Розувастатин противопоказан при беременности и в период грудного вскармливания. Женщины репродуктивного возраста должны применять адекватные методы контрацепции на фоне лечения Розувастатином. Поскольку холестерин и другие продукты биосинтеза холестерина важны для развития плода, потенциальный риск ингибирования ГМГ-КоА редуктазы превышает пользу от применения препарата у беременных. При диагностировании беременности в процессе терапии, прием препарата Розувастатин должен быть прекращен немедленно. Данные в отношении выделения розувастатина с грудным молоком отсутствуют, поэтому в период грудного кормления прием препарата необходимо прекратить. Применение и дозы Внутрь, не разжевывать и не разламывать, проглатывать целиком, запивая водой. Препарат может назначаться в любое время суток независимо от времени приема пищи. До начала терапии препаратом Розувастатин пациент должен начать соблюдать стандартную гипохолестеринемическую диету и продолжать соблюдать ее во время лечения. Доза препарата должна подбираться индивидуально в зависимости от целей терапии и терапевтического ответа на лечение, принимая во внимание текущие рекомендации по целевой концентрации липидов. Рекомендуемая начальная доза для пациентов, начинающих принимать препарат, или для пациентов, переведенных с приема других ингибиторов ГМГ-КоА редуктазы, должна составлять 5 или 10 мг препарата Розувастатин 1 раз в сутки. При выборе начальной дозы следует руководствоваться индивидуальной концентрацией холестерина и принимать во внимание возможный риск сердечно-сосудистых осложнений, а также необходимо оценивать потенциальный риск развития побочных эффектов. В случае необходимости, доза может быть увеличена до большей через 4 недели. В связи с возможным развитием побочных эффектов при приёме дозы 40 мг, по сравнению с более низкими дозами препарата, увеличение дозы до 40 мг, после дополнительного приема дозы выше рекомендуемой начальной дозы в течение 4-х недель терапии, может проводиться только у пациентов с тяжелой степенью гиперхолестеринемии и с высоким риском сердечно-сосудистых осложнений (особенно у пациентов с семейной гиперхолестеринемией), у которых не был достигнут желаемый результат терапии при приёме дозы 20 мг, и которые будут находиться под наблюдением специалиста. Рекомендуется особенно тщательное наблюдение за пациентами, получающими препарат в дозе 40 мг. Не рекомендуется назначение дозы 40 мг пациентам, ранее не обращавшимся к врачу. После 2-4-х недель терапии и/или при повышении дозы препарата Розувастатин необходим контроль показателей липидного обмена (при необходимости требуется коррекция дозы). Пожилые пациенты Начальная рекомендуемая доза у пациентов в возрасте старше 70 лет - 5 мг, в остальном не требуется коррекции дозы в зависимости от возраста. Пациенты с почечной недостаточностью У пациентов с почечной недостаточностью легкой или средней степени тяжести коррекция дозы не требуется. Противопоказано применение всех дозировок препарата Розувастатина у пациентов с тяжелой почечной недостаточностью (КК менее 30 мл/мин.) (см. раздел "Противопоказания"). Противопоказано применение препарата в дозировке 40 мг пациентам с умеренными нарушениями функции почек (КК менее 60 мл/мин.). Пациентам с умеренными нарушениями функции почек (КК менее 60 мл/мин.) рекомендуется начальная доза препарата 5 мг. Пациенты с печеночной недостаточностью Препарат Розувастатин противопоказан пациентам с заболеваниями печени в активной фазе. Особые популяции. Этнические группы При изучении фармакокинетических параметров розувастатина у пациентов, принадлежащих к разным этническим группам, отмечено увеличение системной концентрации розувастатина среди японцев и китайцев. Следует учитывать данный факт при назначении препарата Розувастатин данным группам пациентов. При назначении доз 10 и 20 мг рекомендуемая начальная доза для пациентов монголоидной расы составляет 5 мг. Противопоказано назначение препарата в дозе 40 мг пациентам монголоидной расы (см. раздел "Противопоказания"). Пациенты, предрасположенные к миопатии Противопоказано назначение препарата в дозе 40 мг пациентам с факторами, которые могут указывать на предрасположенность к развитию миопатии (см. раздел "Противопоказания"). При назначении доз 10 и 20 мг рекомендуемая начальная доза для данной группы пациентов составляет 5 мг. Генетический полиморфизм У носителей генотипов SLCOIBI (ОАТР1В1) с.521СС и ABCG2 (BCRP) с.421АА отмечалось увеличение экспозиции (AUC) к розувастатину по сравнению с носителями генотипов SLCOIBI с.52ITT и ABCG2 с.421СС. Для пациентов-носителей генотипов с.521СС или с.421АА рекомендуемая максимальная доза препарата Розувастатин составляет 20 мг 1 раз в сутки. Сопутствующая терапия Розувастатин связывается с различными транспортными белками (в частности, с ОАТР1В1 и BCRP). При одновременном применении препарата Розувастатин с лекарственными препаратами (такими как циклоспорин, некоторые ингибиторы протеазы ВИЧ, включая комбинацию ритонавира с атазанавиром, лопинавиром и/или типранавиром), повышающими концентрацию розувастатина в плазме крови за счет взаимодействия с транспортными белками, может повышаться риск миопатии (включая рабдомиолиз). Следует ознакомиться с инструкцией по применению этих препаратов перед их назначением одновременно с препаратом Розувастатин. В таких случаях следует оценить возможность назначения альтернативной терапии или временного прекращения приема препарата Розувастатин. Если же применение указанных выше препаратов необходимо, следует оценить соотношение пользы и риска сопутствующей терапии препаратом Розувастатин и рассмотреть возможность снижения его дозы (см. раздел "Взаимодействие с другими лекарственными средствами"). Побочные эффекты и передозировка Побочные эффекты: Побочные эффекты, наблюдаемые при приеме розувастатина, обычно выражены незначительно и проходят самостоятельно. Как и при применении других ингибиторов ГМГ-КоА-редуктазы, частота возникновения побочных эффектов носит дозозависимый характер. Частота возникновения нежелательных эффектов представлена следующим образом, согласно классификации Всемирной организации здравоохранения: Часто (>: 1/100, <: 1/10): Нечасто (>: 1/1000, <: 1/100): Редко (>: 1/10000): Очень редко (<: 1/10000), включая отдельные сообщения: Частота не установлена быть подсчитана по имеющимся данным). Иммунная система Редко: реакции повышенной чувствительности, включая ангионевротический отек. Со стороны эндокринной системы Часто: сахарный диабет 2 типа. Со стороны центральной нервной системы Часто: головная боль, головокружение. Со стороны пищеварительного тракта Часто: запор, тошнота, боли в животе. Редко: панкреатит. Со стороны кожных покровов Нечасто: кожный зуд, сыпь, крапивница Со стороны опорно-двигательного аппарата Часто: миалгия. Редко: миопатия (включая миозит), рабдомиолиз. Прочие Часто: астенический синдром. Со стороны мочевыводящей системы У пациентов, получавших розувастатин, может выявляться протеинурия. Изменения количества белка в моче (от отсутствия или следовых количеств до + или больше) наблюдаются у менее 1% пациентов, получающих 10-20 мг препарата, и у приблизительно 3% пациентов, получающих 40 мг препарата. Незначительное изменение количества белка в моче отмечалось при приеме дозы 20 мг. В большинстве случаев протеинурия уменьшается или исчезает в процессе терапии и не означает возникновения острого или прогрессирования существующего заболевания почек. Со стороны опорно-двигательного аппарата При применении розувастатина во всех дозировках и, в особенности при приёме доз препарата, превышающих 20 мг, сообщалось о следующих воздействиях на опорно-двигательный аппарат:миалгия, миопатия (включая миозит), в редких случаях рабдомиолиз с острой почечной недостаточностью или без нее. Дозозависимое повышение активности креатинфосфокиназы (КФК) наблюдалось у незначительного числа пациентов, принимавших розувастатин. В большинстве случаев оно было незначительным, бессимптомным и временным. В случае повышения активности КФК (более чем в 5 раз по сравнению с верхней границей нормы) терапия должна быть приостановлена. Со стороны печени При применении розувастатина наблюдается дозозависимое повышение активности "печеночных" трансаминаз у незначительного числа пациентов. В большинстве случаев оно незначительно, бессимптомно и временно. Лабораторные показатели Повышение концентрации глюкозы, билирубина, активности гамма-глутамилтранспептидазы, щелочной фосфатазы, нарушения функции щитовидной железы. Постмаркетинговое применение Сообщалось о следующих побочных эффектах в постмаркетинговом применении розувастатина: Со стороны системы кроветворения: Частота не установлена: тромбоцитопения. Со стороны пищеварительного тракта Очень редко: желтуха, гепатит. Редко: повышение активности "печеночных" трансаминаз. Частота не установлена: диарея. Со стороны опорно-двигательного аппарата Очень редко: артралгия. Частота не установлена: иммуноопосредованная некротизирующая миопатия. Со стороны центральной нервной системы Очень редко: потеря или снижение памяти. Частота не установлена: периферическая нейропатия. Со стороны дыхательной системы Частота не установлена: кашель, одышка. Со стороны мочевыводящей системы Очень редко: гематурия. Со стороны кожных покровов и подкожно-жировой клетчатки Частота не установлена: синдром Стивенса-Джонсона. Со стороны репродуктивной системы Очень редко: гинекомастия. Прочие Частота не установлена: периферические отеки. При применении некоторых статинов сообщалось о следующих побочных эффектах: депрессия, нарушения сна, включая бессонницу и "кошмарные" сновидения сексуальная дисфункция, гипергликемия, повышение концентрации гликозилированного гемоглобина. Сообщалось о единичных случаях интерстициального заболевания легких, особенно при длительном применении препаратов. Передозировка: Симптомы: при одновременном приеме нескольких суточных доз фармакокинетические параметры розувастатина не изменяются. Лечение: специфического лечения при передозировке розувастатином не существует. При передозировке рекомендуется проводить симптоматическое лечение и мероприятия, направленные на поддержание функций жизненно важных органов и систем. Необходим контроль функции печени и уровня КФК. Маловероятно, что гемодиализ будет эффективен. Взаимодействие с другими ЛС: Ингибиторы транспортных белков Розувастатин связывается с некоторыми транспортными белками, в частности, с ОАТР1В1 и BCRP. Сопутствующее применение препаратов, которые являются ингибиторами транспортных белков, может сопровождаться увеличением концентрации розувастатина в плазме крови и повышенным риском развития миопатии. Циклоспорин: при одновременном применении розувастатина и циклоспорина AUC розувастатина была в среднем в 7 раз выше значения, которое отмечалось у здоровых добровольцев. Не влияет на плазменную концентрацию циклоспорина. Препарат Розувастатин противопоказан пациентам, принимающим циклоспорин (см. раздел "Противопоказания"). Непрямые антикоагулянты: начало терапии розувастатином или увеличение дозы препарата у пациентов, получающих одновременно антагонисты витамина К (например, варфарин), может приводить к увеличению протромбинового времени (Международного Нормализованного Отношения - МНО). Отмена розувастатина или снижение дозы препарата может приводить к уменьшению МНО. В таких случаях рекомендуется контроль МНО. Гемфиброзил и другие гиполипидемические средства: совместное применение розувастатина и гемфиброзила приводит к увеличению в 2 раза максимальной концентрации розувастатина в плазме крови и AUC розувастатина. Основываясь на данных по специфическому взаимодействию, не ожидается фармакокинетически значимого взаимодействия с фенофибратом, возможно фармакодинамическое взаимодействие. Гемфиброзил, фенофибрат, другие фибраты и липидснижающие дозы никотиновой кислоты (более 1 г/сутки) увеличивали риск возникновения миопатии при одновременном применении с ингибиторами ГМГ-КоА-редуктазы, возможно в связи с тем, что они могут вызывать миопатию и при применении в монотерапии. При одновременном приёме препарата с гемфиброзилом, фибратами, никотиновой кислотой в липидснижающих дозах (более 1 г/сутки) пациентам рекомендуется начальная доза препарата 5 мг, прием в дозе 40 мг противопоказан при одновременном назначении с фибратами (см. раздел "Противопоказания"). Эзетимиб: одновременное применение розувастатина в дозе 10 мг и эзетимиба в дозе 10 мг сопровождалось увеличением AUC розувастатина у пациентов с гиперхолестеринемией (см. таблицу 3). Нельзя исключить увеличение риска возникновения побочных эффектов из-за фармакодинамического взаимодействия между препаратом Розувастатин и эзетимибом. Ингибиторы протеазы ВИЧ (вирус иммунодефицита человека): несмотря на то, что точный механизм взаимодействия неизвестен, совместный приём ингибиторов протеазы ВИЧ может приводить к значительному увеличению экспозиции розувастатина (см. таблицу 3). Одновременный прием розувастатина и ингибиторов протеазы ВИЧ при лечении пациентов с ВИЧ не рекомендуется. Антациды: одновременное применение розувастатина и суспензий антацидов, содержащих алюминий и магния гидроксид, приводит к снижению плазменной концентрации розувастатина примерно на 50%. Данный эффект выражен слабее, если антациды применяются через 2 часа после приема розувастатина. Клиническое значение подобного взаимодействия не изучалось. Эритромицин: одновременное применение розувастатина и эритромицина приводит к уменьшению AUC розувастатина на 20% и Сmах розувастатина на 30%. Подобное взаимодействие может возникать в результате усиления моторики кишечника, вызываемого приёмом эритромицина. Пероральные контрацептивы/гормонозаместительная терапия: одновременное применение розувастатина и пероральных контрацептивов увеличивает AUC этинилэстрадиола и AUC норгестрела на 26% и 34% соответственно. Такое увеличение плазменной концентрации должно учитываться при подборе дозы пероральных контрацептивов. Фармакокинетические данные по одновременному применению препарата Розувастатина и гормонозаместительной терапии отсутствуют следовательно, нельзя исключить аналогичного эффекта и при применении данного сочетания. Однако подобная комбинация широко применялась во время проведения клинических исследований и хорошо переносилась пациентами. Другие лекарственные средства: не ожидается клинически значимого взаимодействия розувастатина с дигоксином. Изоферменты цитохрома Р450: результаты исследований in vivo и in vitro показали, что розувастатин не является ни ингибитором, ни индуктором изоферментов цитохрома Р450. Кроме того, розувастатин является слабым субстратом для этих изоферментов. Поэтому не ожидается взаимодействия розувастатина с другими лекарственными средствами на уровне метаболизма с участием изоферментов цитохрома Р450. Не отмечено клинически значимого взаимодействия между розувастатином и флуконазолом (ингибитором изоферментов CYP2C9 и CYP3A4) и кетоконазолом (ингибитором изоферментов CYP2A6 CYP3A4). Взаимодействие с лекарственными средствами, которое требует коррекции дозы розувастатина (см. таблицу 1) Дозу препарата Розувастатин следует корректировать при необходимости его одновременного применения с лекарственными средствами, увеличивающими экспозицию к розувастатину. Если ожидается увеличение экспозиции в 2 раза и более, начальная доза препарата Розувастатин должна составлять 5 мг 1 раз в сутки. Также следует корректировать максимальную суточную дозу препарата Розувастатин так, чтобы ожидаемая экспозиция к розувастатину, не превышала таковую для дозы 40 мг, принимаемой без одновременного назначения лекарственных средств, взаимодействующих с розувастатином. Например, максимальная суточная доза препарата Розувастатин при одновременном применении с гемфиброзилом составляет 20 мг (увеличение экспозиции в 1,9 раза), с ритонавиром/атазанавиром - 10 мг (увеличение экспозиции в 3,1 раза). Таблица 1. Влияние сопутствующей терапии на экспозицию к розувастатину (AUC, данные приведены в порядке убывания)ТАБЛИЦЪ Фармакологическое действие и фармакокинетика Механизм действия Розувастатин является селективным, конкурентным ингибитором ГМГ-КоА-редуктазы, фермента, превращающего 3-гидрокси-3-метилглутарил коэнзим А в мевалонат, предшественник холестерина. Основной мишенью действия розувастатина является печень, где осуществляется синтез холестерина (ХС) и катаболизм липопротеинов низкой плотности (ЛПНП). Розувастатин увеличивает число “печеночных” рецепторов ЛПНП на поверхности клеток, повышая захват и катаболизм ЛПНП, что в свою очередь приводит к ингибированию синтеза липопротеинов очень низкой плотности (ЛПОНП), уменьшая тем самым общее количество ЛПНП и ЛПОНП. Фармакодинамика Розувастатин снижает повышенную концентрацию холестерина-ЛПНП (ХС-ЛПНП), общего холестерина, триглицеридов (ТГ), повышает концентрацию холестерина - липопротеинов высокой плотности (ХС-ЛПВП), а также снижает концентрацию аполипопротеина В (АпоВ), ХС-неЛПВП, ХС-ЛПОНП, ТГ-ЛПОНП и увеличивает уровень аполипопротеина A-I (АпоА-I), снижает соотношение ХС-ЛПНП/ХС-ЛПВП, общий ХС/ХС-ЛПВП и ХС-неЛПВП/ ХС-ЛПВП и соотношение АпоВ/АпоА-1. возможность снижениект появляется в течение одной недели после начала терапии препаратом Розувастатин, через 2 недели лечения достигает 90% от максимально возможного эффекта. Максимальный терапевтический эффект обычно достигается к 4-ой неделе и поддерживается при регулярном приёме препарата. Клиническая эффективность Розувастатин эффективен у взрослых пациентов с гиперхолестеринемией с или без гипертриглицеридемии: вне зависимости от расовой принадлежности, пола или возраста, в т.ч. у пациентов с сахарным диабетом и семейной гиперхолестеринемией. У 80% пациентов с гиперхолестеринемией IIа и IIb типа по классификации Фредриксона (средняя исходная концентрация ХС-ЛПНП около 4,8 ммоль/л) на фоне приема препарата в дозе 10 мг концентрация ХС-ЛПНП достигает значений менее 3 ммоль/л. У пациентов с гетерозиготной семейной гиперхолестеринемией, получающих розувастатин в дозе 20-80 мг, отмечается положительная динамика показателей липидного профиля. После титрования до суточной дозы 40 мг (12 недель терапии), отмечается снижение концентрации ХС-ЛПНП на 53%. У пациентов с гомозиготной семейной гиперхолестеринемией, принимающих розувастатин в дозе 20 мг и 40 мг, среднее снижение концентрации ХС-ЛПНП составляет 22%. Аддитивный эффект отмечается в комбинации с фенофибратом в отношении концентрации триглицеридов и с никотиновой кислотой в липидснижающих дозах (более 1 г/сутки) в отношении концентрации ХС-ЛПВП. Фармакокинетика: Абсорбция и распределение Максимальная концентрация розувастатина в плазме крови достигается приблизительно через 5 часов после приема внутрь. Абсолютная биодоступность составляет примерно 20 %. Розувастатин метаболизируется преимущественно печенью, которая является основным местом синтеза холестерина и метаболизма ХС-ЛПНП. Объем распределения розувастатина составляет примерно 134 л. Приблизительно 90% розувастатина связывается с белками плазмы крови, в основном с альбумином. Метаболизм Подвергается ограниченному метаболизму (около 10%). Розувастин является непрофильным субстратом для метаболизма изоферментами системы цитохрома Р450. Основным изоферментом, участвующим в метаболизме розувастатина, является изофермент CYP2C9. Изоферменты CYP2C19, CYP3A4 и CYP2D6 вовлечены в метаболизм в меньшей степени. Основными выявленными метаболитами розувастатина являются N-дисметил-метаболит и лактоновые метаболиты. N-дисметил примерно на 50% менее активен, чем розувастатин, лактоновые метаболиты фармакологически не активны. Более 90% фармакологической активности по ингибированию циркулирующей ГМГ-КоА редуктазы обеспечивается розувастатином, остальное - его метаболитами. Выведение Около 90% дозы розувастатина выводится в неизмененном виде через кишечник (включая абсорбированный и не абсорбированный розувастатин). Оставшаяся часть выводится почками. Плазменный период полувыведения (Т1/2) составляет примерно 19 часов. Т1/2, не изменяется при увеличении дозы препарата. Средний геометрический плазменный клиренс составляет приблизительно 50 л/ч (коэффициент вариации 21,7%). Как и в случае других ингибиторов ГМГ-КоА редуктазы, в процесс “печеночного” захвата розувастатина вовлечен мембранный переносчик холестерина, выполняющий важную роль в печеночной элиминации розувастатина. Линейность Системная экспозиция розувастатина увеличивается пропорционально дозе. Фармакокинетические параметры не изменяются при ежедневном приеме. Особые популяции пациентов Возраст и пол Пол и возраст не оказывают клинически значимого влияния на фармакокинетику розувастатина. Этнические группы Фармакокинетические исследования показали приблизительно двукратное увеличение медианы AUC (площади под кривой "концентрация-время") и Сmах (максимальной концентрации в плазме крови) розувастатина у пациентов азиатской национальности (японцев, китайцев, филиппинцев, вьетнамцев и корейцев) по сравнению с европейцами: у индийских пациентов показано увеличение медианы AUC и Сmах в 1,3 раза. Фармакокинетический анализ не выявил клинически значимых различий в фармакокинетике среди европейцев и представителей негроидной расы. Почечная недостаточность У пациентов с легкой и умеренно выраженной почечной недостаточностью величина плазменной концентрации розувастатина или N-дисметил-метаболита существенно не меняется. У пациентов с тяжелой почечной недостаточностью (клиренс креатинина (КК) менее 30 мл/мин) концентрация розувастатина в плазме крови в 3 раза выше, а концентрация N-дисметил-метаболита в 9 раз выше, чем у здоровых добровольцев. Концентрация розувастатина в плазме крови у пациентов на гемодиализе была примерно на 50% выше, чем у здоровых добровольцев. Печеночная недостаточность У пациентов с различной степенью печеночной недостаточности не выявлено увеличение Т1/2 розувастатина у пациентов с баллом 7 и ниже по шкале Чайлд-Пью. У двух пациентов с баллами 8 и 9 по шкале Чайлд-Пью отмечено увеличение Т1/2, по крайней мере, в 2 раза. Опыт применения розувастатина у пациентов с баллом выше 9 по шкале Чайлд-Пью отсутствует. Генетический полиморфизм Ингибиторы ГМГ-КоА-редуктазы, в том числе и розувастатин, связываются с транспортными белками ОАТР1В1 (полипептид транспорта органических анионов, участвующих в захвате статинов гепатоцитами) и BCRP (эффлюксный транспортер). У носителей генотипов SLCOIBI (ОАТР1В1) с.521СС и ABCG2 (BCRP) с.421АА отмечалось увеличение экспозиции (AUC) к розувастатину в 1,6 и 2,4 раза, соответственно, по сравнению с носителями генов SLCOIBI с.52ITT и ABCG2 с.421СС. Особые указания Со стороны мочевыделительной системы У пациентов, получавших высокие дозы розувастатина (в основном 40 мг), наблюдалась канальцевая протеинурия, которая в большинстве случаев была транзиторной. Такая протеинурия не свидетельствовала об остром заболевании почек или прогрессировании заболевания почек. У пациентов, принимающих препарат в дозе 40 мг, рекомендуется контролировать показатели функции почек во время лечения. Со стороны опорно-двигательного аппарата При применении розувастатина во всех дозировках и, в особенности при приёме доз препарата, превышающих 20 мг, сообщалось о следующих воздействиях на опорно-двигательный аппарат: миалгия, миопатия, в редких случаях рабдомиолиз. Определение активности креатинфосфокиназы Определение активности КФК не следует проводить после интенсивных физических нагрузок или при наличии других возможных причин увеличения активности КФК, что может привести к неверной интерпретации полученных результатов. В случае если исходная активность КФК существенно повышена (в 5 раз выше, чем верхняя граница нормы), через 5-7 дней следует провести повторное измерение. Не следует начинать терапию, если повторный тест подтверждает исходную активность КФК (выше более чем в 5 раз по сравнению с верхней границей нормы). До начала терапии При применении препарата Розувастатин, также как и других ингибиторов ГМГ-КоА- редуктазы, следует проявлять осторожность пациентам с имеющимися факторами риска миопатии/рабдомиолиза, необходимо рассмотреть соотношение риска и возможной пользы терапии и проводить клиническое наблюдение. Во время терапии Следует проинформировать пациента о необходимости немедленного сообщения врачу о случаях неожиданного появления мышечных болей, мышечной слабости или спазмах, особенно в сочетании с недомоганием и лихорадкой. У таких пациентов следует определять активность КФК. Терапия должна быть прекращена если активность КФК значительно увеличена (более чем в 5 раз по сравнению с верхней границей нормы) или если симптомы со стороны мышц резко выражены и вызывают ежедневный дискомфорт (даже, если активность КФК в 5 раз меньше по сравнению с верхней границей нормы). Если симптомы исчезают, и активность КФК возвращается к норме, следует рассмотреть вопрос о повторном применении препарата Розувастатин или других ингибиторов ГМГ-КоА-редуктазы в меньших дозах при тщательном наблюдении за пациентом. Рутинный контроль активности КФК при отсутствии симптомов нецелесообразен. Отмечены очень редкие случаи иммуноопосредованной некротизирующей миопатии с клиническими проявлениями в виде стойкой слабости проксимальных мышц и повышения активности КФК в сыворотке во время лечения или при прекращении приема статинов, в т.ч. розувастатина. Может потребоваться проведение дополнительных исследований мышечной системы и нервной системы, серологических исследований, а также терапия иммунодепрессивными средствами. Не отмечено признаков увеличения воздействия на скелетную мускулатуру при приеме розувастатин и сопутствующей терапии. Однако сообщалось об увеличении числа случаев миозита и миопатии у пациентов, принимавших другие ингибиторы ГМГ-КоА-редуктазы в сочетании с производными фибриновой кислоты, включая гемфиброзил, циклоспорин, никотиновую кислоту в липидснижающих дозах (более 1 г/сутки), азольные противогрибковые средства, ингибиторы протеазы ВИЧ и макролидные антибиотики. Гемфиброзил увеличивает риск возникновения миопатии при совместном применении с некоторыми ингибиторами ГМГ-КоА-редуктазы. Таким образом, не рекомендуется одновременное применение препарата Розувастатин и гемфиброзила. Следует тщательно взвесить соотношение риска и возможной пользы при совместном применении препарата Розувастатин и фибратов или липидснижающих доз никотиновой кислоты (более 1 г/сутки). Противопоказан прием препарата Розувастатин в дозе 40 мг совместно с фибратами. Через 2-4 недели после начала лечения и/или при повышении дозы препарата Розувастатин необходим контроль показателей липидного обмена (при необходимости требуется коррекция дозы). Влияние на функцию печени Рекомендуется проводить определение показателей функции печени до начала терапии и через 3 месяца после начала терапии. Прием препарата Розувастатин следует прекратить или уменьшить дозу препарата, если активность "печеночных" трансаминаз в сыворотке крови в 3 раза превышает верхнюю границу нормы. У пациентов с гиперхолестеринемией вследствие гипотиреоза или нефротического синдрома терапия основных заболеваний должна проводиться до начала лечения препаратом Розувастатин. Особые популяции. Этнические группы В ходе фармакокинетических исследований среди китайских и японских пациентов отмечено увеличение системной концентрации розувастатина по сравнению с показателями, полученными среди европейцев. Ингибиторы протеазы ВИЧ Не рекомендуется совместное применение препарата с ингибиторами протеазы ВИЧ. Лактоза Препарат не следует применять у пациентов с лактозной недостаточностью, непереносимостью лактазы и глюкозо-галактозной мальабсорбцией. Интерстициальное заболевание легких При применении некоторых статинов особенно в течение длительного времени сообщалось о единичных случаях интерстициального заболевания легких. Проявлениями заболевания могут являться одышка, непродуктивный кашель и ухудшение, общего самочувствия (слабость, снижение веса и лихорадка). При подозрении на интерстициальное заболевание легких следует прекратить терапию статинами. Сахарный диабет 2 типа У па
634 Руб.Розувастатин СЗ таб. п/о 20мг №90

Показания Первичная гиперхолестеринемия по классификации Фредриксона (тип IIа, включая семейную гетерозиготную гиперхолестеринемию) или смешанная гиперхолестеринемия (тип IIb) в качестве дополнения к диете, когда диета и другие немедикаментозные методы лечения (например, физические упражнения, снижение массы тела) оказываются недостаточными- Семейная гомозиготная гиперхолестеринемия в качестве дополнения к диете и другой липидснижающей терапии (например, ЛПНП-аферез), или в случаях, когда подобная терапия недостаточно эффективна- Гипертриглицеридемия (тип IV по классификации Фредриксона) в качестве дополнения к диете- Для замедления прогрессирования атеросклероза в качестве дополнения к диете у пациентов, которым показана терапия для снижения концентрации общего ХС и ХС-ЛПНП- Первичная профилактика основных сердечно-сосудистых осложнений (инсульта, инфаркта, артериальной реваскуляризации) у взрослых пациентов без клинических признаков ИБС, но с повышенным риском ее развития (возраст старше 50 лет для мужчин и старше 60 лет для женщин, повышенная концентрация С-реактивного белка (более 2 мг/л) при наличии, как минимум одного из дополнительных факторов риска, таких как артериальная гипертензия, низкая концентрация ХС-ЛПВП, курение, семейный анамнез раннего начала ИБС). Противопоказания Противопоказания Для препарата Розувастатин-СЗ в суточной дозе 5 мг, 10 мг и 20 мг- повышенная чувствительность к розувастатину или любому из компонентов препарата- непереносимость лактозы, дефицит лактазы или глюкозо-галактозная мальабсорбция (препарат содержит лактозу)- детский возраст до 18 лет- заболевания печени в активной фазе, включая стойкое повышение сывороточной активности трансаминаз и любое повышение активности трансаминаз в сыворотке крови (более чем в 3 раза по сравнению с верхней границей нормы)- тяжелая почечная недостаточность (КК менее 30 мл/мин.)- миопатия- одновременный прием циклоспорина- у женщин: беременность: период грудного вскармливания, отсутствие адекватных методов контрацепции- повышение концентрации креатинфосфокиназы (КФК) в крови более чем в 5 раз по сравнению с верхней границей нормы- совместное применение с ингибиторами ВИЧ-протеаз- пациентам, предрасположенным к развитию миотоксических осложнений. Для препарата Розувастатин-СЗ в суточной дозе 40 мг- повышенная чувствительность к розувастатину или любому из компонентов препарата- непереносимость лактозы, дефицит лактазы или глюкозо-галактозная мальабсорбция (препарат содержит лактозу)- детский возраст до 18 лет- одновременный прием циклоспорина- у женщин: беременность, период грудного вскармливания, отсутствие адекватных методов контрацепции- повышение концентрации креатинфосфокиназы (КФК) в крови более чем в 5 раз по сравнению с верхней границей нормы- совместное применение с ингибиторами ВИЧ-протеаз- почечная недостаточность средней и тяжелой степени (КК менее 60 мл/мин.)- заболевания печени в активной фазе, включая стойкое повышение сывороточной активности трансаминаз и любое повышение активности трансаминаз в сыворотке крови (более чем в 3 раза по сравнению с верхней границей нормы) пациентам с факторами риска развития миопатии/рабдомиолиза, а именно- гипотиреоз- миотоксичность на фоне приема других ингибиторов ГМГ-КоА-редуктазы или фибратов в анамнезе- чрезмерное употребление алкоголя- состояния, которые могут приводить к повышению плазменной концентрации розувастатина- одновременный прием фибратов- миопатия- личный или семейный анамнез мышечных заболеваний - пациентам монголоидной расы С осторожностью: Для препарата Розувастатин-СЗ в суточной дозе 5 мг, 10 мг и 20 мг: Наличие риска развития миопатии/рабдомиолиза - почечная недостаточность, гипотиреоз, личный или семейный анамнез наследственных мышечных заболеваний и предшествующий анамнез мышечной токсичности при использовании других ингибиторов ГМГ-КоА-редуктазы или фибратов: чрезмерное употребление алкоголя: возраст старше 65 лет: состояния, при которых отмечено повышение плазменной концентрации розувастатина: расовая принадлежность (монголоидная раса): одновременное назначение с фибратами (см. раздел "Фармакокинетика"): заболевания печени в анамнезе: сепсис: артериальная гипотензия: обширные хирургические вмешательства, травмы, тяжелые метаболические, эндокринные или электролитные нарушения или неконтролируемые судорожные припадки. Одновременное применение с колхицином и с эзетимибом (см. раздел "Взаимодействие с другими лекарственными препаратами"). Для препарата Розувастатин-СЗ в суточной дозе 40 мг: Почечная недостаточность легкой степени тяжести (КК более 60 мл/мин): возраст старше 65 лет: заболевания печени в анамнезе: сепсис: артериальная гипотензия: обширные хирургические вмешательства, травмы, тяжелые метаболические, эндокринные или электролитные нарушения или неконтролируемые судорожные припадки. Одновременное применение с колхицином и с эзетимибом (см. раздел "Взаимодействие с другими лекарственными препаратами"). Пациенты с печеночной недостаточностью Данные или опыт применения препарата у пациентов с более чем 9-ю баллами по шкале Чайлд-Пью отсутствует (см. разделы "Фармакодинамика" и "Особые указания"). Беременность Розувастатин-СЗ противопоказан при беременности и в период грудного вскармливания. Женщины репродуктивного возраста должны применять адекватные методы контрацепции. Поскольку холестерин и другие продукты биосинтеза холестерина важны для развития плода, потенциальный риск ингибирования ГМГ-КоА-редуктазы превышает пользу от применения препарата у беременных. В случае возникновения беременности в процессе терапии прием препарата должен быть прекращен немедленно. Данные в отношении выделения розувастатина с грудным молоком отсутствуют, поэтому в период грудного вскармливания прием препарата необходимо прекратить (см. раздел "Противопоказания"). Применение и дозы Внутрь, не разжевывать и не измельчать таблетку, проглатывать целиком, запивая водой. Препарат может назначаться в любое время суток независимо от приема нищи. До начала терапии препаратом Розувастатин-СЗ пациент должен начать соблюдать стандартную гипохолестеринемическую диету и продолжать соблюдать ее во время лечения. Доза препарата должна подбираться индивидуально в зависимости от целей терапии и терапевтического ответа на лечение, принимая во внимание текущие рекомендации по целевым концентрациям липидов. Рекомендуемая начальная доза для пациентов, начинающих принимать препарат, или для пациентов, переведенных с приема других ингибиторов ГМГ-КоА-редуктазы, должна составлять 5 мг или 10 мг препарата Розувастатин-СЗ 1 раз в сутки. При выборе начальной дозы следует руководствоваться индивидуальным содержанием холестерина и принимать во внимание возможный риск сердечнососудистых осложнений, а также необходимо оценивать потенциальный риск развития побочных эффектов. В случае необходимости, доза может быть увеличена до большей через 4 недели (см. раздел "Фармакодинамика").В связи с возможным развитием побочных эффектов при приеме дозы 40 мг, по сравнению с более низкими дозами препарата (см. раздел "Побочное действие"), увеличение дозы до 40 мг, после дополнительного приема дозы выше рекомендуемой начальной дозы в течение 4-х недель терапии, может проводиться только у пациентов с тяжелой степенью гиперхолестеринемии и с высоким риском сердечно-сосудистых осложнений (особенно у пациентов с семейной гиперхолестеринемией), у которых не был достигнут желаемый результат терапии при приеме дозы 20 мг, и которые будут находиться под наблюдением специалиста (см. раздел "Особые указания"). Рекомендуется особенно тщательное наблюдение за пациентами, получающими препарат в дозе 40 мг. Не рекомендуется назначение дозы 40 мг пациентам, ранее не обращавшимся к врачу. После 2-4-х недель терапии и/или при повышении дозы препарата Розувастатин-СЗ необходим контроль показателей липидного обмена (при необходимости требуется коррекция дозы). Пожилые пациенты. Не требуется коррекции дозы. Пациенты с почечной недостаточностьюУ пациентов с почечной недостаточностью легкой или средней степени тяжести коррекция дозы не требуется. У пациентов с тяжелой почечной недостаточностью (КК менее 30 мл/мин.) применение препарата Розувастатин-СЗ противопоказано. Противопоказано применение препарата в дозе 40 мг пациентам с умеренными нарушениями функции почек (КК 30-60 мл/мин.) (см. разделы "Особые указания" и "Фармакодинамика"). Пациентам с умеренными нарушениями функции почек рекомендуется начальная доза препарата 5 мг. Пациенты с печеночной недостаточностью Розувастатин-СЗ противопоказан пациентам с заболеваниями печени в активной фазе (см. раздел "Противопоказания"). Особые популяции. Этнические группы При изучении фармакокинетических параметров розувастатина у пациентов, принадлежащих к разным этническим группам, отмечено увеличение системной концентрации розувастатина среди японцев и китайцев (см. раздел "Особые указания"). Следует учитывать данный факт при назначении препарата Розувастатин-СЗ данным группам пациентов. При назначении доз 10 мг и 20 мг рекомендуемая начальная доза для пациентов монголоидной расы составляет 5 мг. Противопоказано назначение препарата в дозе 40 мг пациентам монголоидной расы (см. раздел "Противопоказания"). Генетический полиморфизм У носителей генотипов SLC01B1 (ОАТР1В1) с.521СС и ABCG2 (BCRP) с.421АА отмечалось увеличение экспозиции (AUC) к розувастатину но сравнению с носителями генотипов SLCOIBI с.521ТТ и ABCG2 с.421СС. Для пациентов-носителей генотипов с.521СС или с.421АА рекомендуемая максимальная доза препарата Розувастатин-СЗ составляет 20 мг один раз в сутки (см. разделы "Фармакокинетика", "Особые указания" и "Взаимодействие с другими лекарственными препаратами"). Пациенты предрасположенные к миопатии Противопоказано назначение препарата в дозе 40 мг пациентам с факторами, которые могут указывать на предрасположенность к развитию миопатии (см. раздел "Противопоказания"). При назначении доз 10 и 20 мг рекомендуемая начальная доза для данной группы пациентов составляет 5 мг (см. раздел "Противопоказания"). Сопутствующая терапия Розувастатин связывается с различными транспортными белками (в частности, с ОАТР1В1 и BCRP). При совместном применении препарата Розувастатин-СЗ с лекарственными препаратами (такими как циклоспорин, некоторые ингибиторы протеазы ВИЧ, включая комбинацию ритонавира с атазанавиром, лопииавиром и/или типранавиром), повышающими концентрацию розувастатина в плазме за счет взаимодействия с транспортными белками, может повышаться риск миопатии (включая рабдомиолиз) (см. разделы "Особые указания" и "Взаимодействие с другими лекарственными препаратами"). В таких случаях следует оценить возможность назначения альтернативной терапии или временного прекращения приема препарата Розувастатин-СЗ. Если же применение указанных выше препаратов необходимо, следует оценить соотношение пользы и риска сопутствующей терапии препаратом Розувастатин-СЗ и рассмотреть возможность снижения его дозы (см. раздел "Взаимодействие с другими лекарственными препаратами"). Побочные эффекты и передозировка Побочные эффекты: Побочные эффекты, наблюдаемые при приеме препарата Розувастатин-СЗ, обычно выражены незначительно и проходят самостоятельно. Как и при применении других ингибиторов ГМГ-КоА-редуктазы, частота возникновения побочных эффектов носит, в основном, дозозависимый характер. Частота возникновения нежелательных эффектов представлена следующим образом: часто (>: 1/100, <:1/10): нечасто (>: 1/1000, <:1/100): редко (>: 1/10000, <: 1/1000): очень редко (<: 1/10000), неуточненная частота (не может быть подсчитана по имеющимся данным). Иммунная система Редко: реакции повышенной чувствительности, включая ангионевротический отек. Эндокринная система Часто: сахарный диабет 2-го типа Со стороны центральной нервной системы Часто: головная боль, головокружение Со стороны пищеварительного тракта. Часто: запор, тошнота, боли в животе. Редко: панкреатит Со стороны кожных покровов Нечасто: кожный зуд, сыпь, крапивница Со стороны опорно-двигательного Редко: панкреатит. Со стороны кожных покровов Нечасто: кожный зуд, сыпь, крапивница Со стороны опорно-двигательного аппарата. При применении препарата Розувастатин- СЗ во всех дозах и, в особенности при приеме доз препарата, превышающих 20 мг, сообщалось о следующих воздействиях на опорно-двигательный аппарат: миалгия, миопатия (включая миозит), в редких случаях - рабдомиолиз с острой почечной недостаточностью или без нее. Дозозависимое повышение активности креатинфосфокиназы (КФК) наблюдается у незначительного числа пациентов, принимавших розувастатин. В большинстве случаев оно было незначительным, бессимптомным и временным. В случае повышения активности КФК (более чем в 5 раз по сравнению с верхней границей нормы) терапия должна быть приостановлена (см. раздел "Особые указания"). Со стороны печени При применении розувастатина наблюдается дозозависимое повышение активности "печеночных" трансаминаз у незначительного числа пациентов. В большинстве случаев оно незначительно, бессимптомно и временно. Лабораторные показатели При применении препарата Розувастатин- СЗ так же наблюдались следующие изменения лабораторных показателей: повышение концентрации глюкозы, билирубина, активности гамма- глутамилтранспептидазы, щелочной фосфатазы, нарушения функции щитовидной железы. Постмаркетинговое применение Сообщалось о следующих побочных эффектах в постмаркетинговом применении препарата Розувастатин-СЗ: Со стороны системы кроветворения Неуточненной частоты: тромбоцитопения Со стороны пищеварительного тракта Очень редко: желтуха, гепатит Редко: повышение активности "печеночных" трансаминаз Неуточненной частоты: диарея. Со стороны опорно-двигательного аппарата. Очень редко: артралгия. Неуточненной частоты: иммуноопосредованная некротизирующая миопатия Со стороны центральной нервной системы Очень редко: полинейропатия, потеря памяти Со стороны дыхательной системы Неуточненной частоты: кашель, одышка Со стороны мочевыводящей системы Очень редко: гематурия. Со стороны кожных покровов и подкожно-жировой клетчатки. Неуточненной частоты: синдром Стивенса- Джонсона. Со стороны репродуктивной системы и молочной железы. Неуточненной частоты: гинекомастия Прочие Неуточненной частоты: периферические отеки. При применении некоторых статинов сообщалось о следующих побочных эффектах: депрессия, нарушения сна, включая бессонницу и "кошмарные" сновидения, сексуальная дисфункция. Сообщалось о единичных случаях интерстициального заболевание легких, особенно при длительном применении препаратов (см. раздел "Особые указания"). Передозировка: При одновременном приеме нескольких суточных доз фармакокинетические параметры розувастатина не изменяются. Специфического лечения при передозировке розувастатином не существует. При передозировке рекомендуется проводить симптоматическое лечение и мероприятия, направленные на поддержание функций жизненно важных органов и систем. Необходим контроль функции печени и уровня КФК. Маловероятно, что гемодиализ будет эффективен. Взаимодействие с другими ЛС: Влияние применения других препаратов на розувастатин Ингибиторы транспортных белков: Розувастатин связывается с некоторыми транспортными белками, в частности, с ОАТР1В1 и BCRP. Сопутствующее применение препаратов, которые являются ингибиторами этих транспортных белков, может сопровождаться увеличением концентрации розувастатина в плазме и повышенным риском развития миопатии (см. таблицу 1 и разделы "Способ применения и дозы" и "Особые указания"). Циклоспорин: при одновременном применении розувастатина и циклоспорина AUC розувастатина была в среднем в 7 раз выше значения, которое отмечалось у здоровых добровольцев (см. таблицу 1). Не влияет на плазменную концентрацию циклоспорина. Розувастатин-СЗ противопоказан пациентам, принимающим циклоспорин (см. раздел "Противопоказания"). Ингибиторы протеазы вируса иммунодефицита человека (ВИЧ): несмотря на то, что точный механизм взаимодействия неизвестен, совместный прием ингибиторов протеазы ВИЧ может приводить к значительному увеличению экспозиции розувастатина (см. таблицу 1). Фармакокинетическое исследование но одновременному применению 20 мг розувастатина с комбинированным препаратом, содержащим два ингибитора протеазы ВИЧ (400 мг лопинавира/100 мг ритонавира) у здоровых добровольцев приводило к приблизительно двукратному и пятикратному увеличению AUC(0-24) и Сmах розувастатина, соответственно. Поэтому одновременный прием розувастатина и ингибиторов протеазы ВИЧ не рекомендуется (см. разделы "Способ применения и дозы", "Противопоказания" и "Особые указания", таблицу 1). Гемфиброзил и другие гиполипидемические средства: совместное применение розувастатина и гемфиброзила приводит к увеличению в 2 раза максимальной концентрации розувастатина в плазме крови и AUC розувастатина (см. раздел "Особые указания"). Основываясь на данных по специфическому взаимодействию, не ожидается фармакокинетически значимого взаимодействия с фенофибратом, возможно фармакодинамическое взаимодействие. Гемфиброзил, фенофибрат, другие фибраты и липидснижающие дозы никотиновой кислоты увеличивали риск возникновения миопатии при одновременном применении с ингибиторами ГМГ-КоА-редукгазы, возможно в связи с тем, что они могут вызывать миопатию при применении в монотерапии (см. раздел "Особые указания"). При одновременном приеме препарата с гемфиброзилом, фибратами, никотиновой кислотой в липидснижающих дозах (более 1 г/сут.) пациентам рекомендуется начальная доза препарата 5 мг, прием в дозе 40 мг противопоказан при совместном назначении с фибратами (см. разделы "Противопоказания", "Способ применения и дозы", "Особые указания"). Эзетимиб: одновременное применение препарата Розувастатин-СЗ в дозе 10 мг и эзетимиба в дозе 10 мг сопровождалось увеличением AUC розувастатина у пациентов с гиперхолестеринемией (см. таблицу 1). Нельзя исключить увеличение риска возникновения побочных эффектов из-за фармакодинамического взаимодействия между препаратом Розувастатин-СЗ и эзетимибом. Антациды: одновременное применение розувастатина и суспензий антацидов, содержащих магния и алюминия гидроксид, приводит к снижению плазменной концентрации розувастатина примерно на 50 %. Данный эффект выражен слабее, если антациды применяются через 2 часа после приема розувастатина. Клиническое значение подобного взаимодействия не изучалось. Эритромицин: одновременное применение розувастатина и эритромицина приводит к уменьшению AUC розувастатина на 20 % и Сmах розувастатина па 30 %. Подобное взаимодействие может возникать в результате усиления моторики кишечника, вызываемого приемом эритромицина. Изоферменты цитохрома Р450: результаты исследований in vivo и in vitro показали, что розувастатин не является ни ингибитором, пи индуктором изоферментов цитохрома Р450. Кроме того, розувастатин является слабым субстратом для этих изоферментов. Поэтому не ожидается взаимодействия розувастатина с другими лекарственными средствами на уровне метаболизма с участием изоферментов цитохрома Р450. Не отмечено клинически значимого взаимодействия розувастатина с флуконазолом (ингибитором изоферментов CYP2C9 и CYP3A4) и кетоконазолом (ингибитором изоферментов CYP2A6 и CYP3A4). Взаимодействие с лекарственными средствами, которое требует коррекции дозы розувастатина (см. таблицу 1) Дозу препарата Розувастатии-СЗ следует корректировать при необходимости его совместного применения с лекарственными средствами, увеличивающими экспозицию к розувастатину. Если ожидается увеличение экспозиции в 2 раза и более, начальная доза препарата Розувастатин-СЗ должна составлять 5 мг один раз в сутки. Также следует корректировать максимальную суточную дозу препарата Розувастатин-СЗ так, чтобы ожидаемая экспозиция к розувастатину не превышала таковую для дозы 40 мг, принимаемой без одновременного назначения лекарственных средств, взаимодействующих с розувастатином. Например, максимальная суточная доза препарата Розувастатин-СЗ при одновременном применении с гемфиброзилом составляет 20 мг (увеличение экспозиции в 1,9 раза), с ритонавиром/атазанавиром - 10 мг (увеличение экспозиции в 3,1 раза). Таблица 1. Влияние сопутствующей терапии на экспозицию к розувастатину (AUC, данные приведены в порядке убывания) - результаты опубликованных клинических исследований убывания) - результаты опубликованных клинических исследований ТАБЛИЦЪ Влияние применения розувастатина на другие препараты Антагонисты витамина К: начало терапии розувастатином или увеличение дозы препарата у пациентов, получающих одновременно антагонисты витамина К (например, варфарин), может приводить к увеличению Международного Нормализованного Отношения (MHO). Отмена розувастатина или снижение дозы препарата может приводить к уменьшению MHO. В таких случаях рекомендуется контроль MHO. Пероральные контрацептивы/ гормонозаместительная терапия: одновременное применение розувастатина и пероральных контрацептивов увеличивает AUC этинилэстрадиола и AUC норгестрела на 26 % и 34 %, соответственно. Такое увеличение плазменной концентрации должно учитываться при подборе дозы пероральных контрацептивов. Фармакокинетические данные по одновременному применению препарата Розувастатин-СЗ и гормонозамести- тельной терапии отсутствуют, следовательно, нельзя исключить аналогичного эффекта и при применении данного сочетания. Однако подобная комбинация широко применялась во время проведения клинических исследований и хорошо переносилась пациентами. Другие лекарственные средства: не ожидается клинически значимого взаимодействия розувастатина с дигоксином. Фармакологическое действие и фармакокинетика Механизм действия Розувастатин является селективным, конкурентным ингибитором ГМГ-КоА- редуктазы, фермента, превращающего З-гидрокси-З-метилглутарил кофермент А в мевалоновую кислоту, предшественник холестерина. Основной мишенью действия розувастатина является печень, где осуществляется синтез холестерина (ХС) и катаболизм липопротеинов низкой плотности (ЛПНП). Розувастатин увеличивает число "печеночных" рецепторов ЛПНП на поверхности клеток, повышая захват и катаболизм ЛПНП, что в свою очередь приводит к ингибированию синтеза липопротеинов очень низкой плотности (ЛПОНП), уменьшая тем самым общее количество ЛПНП и ЛПОНП. Фармакодинамика Розувастатин-СЗ снижает повышенные концентрации холестерина-ЛПНП (ХС-ЛПНП), общего холестерина, триглицеридов (ТГ), повышает концентрацию холестерина-липопротеинов высокой плотности (ХС-ЛПВП), а также снижает концентрации аполипопротеина В (АпоВ), ХС-неЛПВП, ХС-ЛПОНП, ТГ-ЛПОНП и увеличивает концентрацию аполипопротеина A-I (АпоА-1), снижает соотношение ХС-ЛПНП/ХС-ЛПВП, общий ХС/ХС-ЛПВП и ХС-неЛПВП/ХС-ЛПВП и соотношение АпоВ/АпоА-1. Терапевтический эффект развивается в течение одной недели после начала терапии препаратом Розувастатин-СЗ, через 2 недели лечения достигает 90 % от максимально возможного эффекта. Максимальный терапевтический эффект обычно достигается к 4-ой неделе терапии и поддерживается при регулярном приеме препарата. Розувастатин-СЗ эффективен у взрослых пациентов с гиперхолестеринемией с или без гипертриглицеридемии, вне зависимости от расовой принадлежности, пола или возраста, в том числе, у пациентов с сахарным диабетом и семейной гиперхолестеринемией. У 80 % пациентов с гиперхолестеринемией IIа и IIb типа по Фредриксону (средняя исходная концентрация ХС-ЛПНП около 4,8 ммоль/л) на фоне приема препарата в дозе 10 мг концентрация ХС-ЛПНП достигает значений менее 3 ммоль/л. У пациентов с гетерозиготной семейной гиперхолестеринемией, получающих Розувастатин-СЗ в дозе 20-80 мг, отмечается положительная динамика показателей липидного профиля. После титрования до суточной дозы 40 мг (12 недель терапии), отмечается снижение концентрации ХС-ЛПНП на 53 %. У 33 % пациентов достигается концентрация ХС-ЛПНП менее 3 ммоль/л. У пациентов с гомозиготной семейной гиперхолестеринемией, принимающих Розувастатин-СЗ в дозе 20 мг и 40 мг, среднее снижение концентрации ХС-ЛПНП составляет 22 %. У пациентов с гипертриглицеридемией с начальной концентрацией ТГ от 273 до 817 мг/дл, получавших Розувастатин-СЗ в дозе от 5 мг до 40 мг один раз в сутки в течение 6-ти недель, значительно снижалась концентрация ТГ в плазме крови. Аддитивный эффект отмечается в комбинации с фенофибратом в отношении содержания триглицеридов и с никотиновой кислотой в липидснижающих дозах в отношении содержания ХС-ЛПВП (см. также раздел "Особые указания"). По результатам клинических исследований пациентам с выраженной гиперхолестеринемией и высоким риском сердечно-сосудистых заболеваний (ССЗ) должна назначаться доза препарата Розувастатин-СЗ 40 мг. Результаты клинического исследования (Обоснование применения статинов для первичной профилактики: интервенционное исследование по оценке розувастатина) показали, что розувастатин существенно снижал риск развития сердечно-сосудистых осложнений. Фармакокинетика: Абсорбция и распределение Максимальная концентрация розувастатина в плазме крови достигается приблизительно через 5 часов после приема внутрь. Абсолютная биодостунность составляет примерно 20 %. Розувастатин метабол изируется преимущественно печенью, которая является основным местом синтеза холестерина и метаболизма ХС-ЛПНП. Объем распределения розувастатина составляет примерно 134 л. Приблизительно 90 % розувастатина связывается с белками плазмы крови, в основном с альбумином. Метаболизм Подвергается ограниченному метаболизму (около 10 %). Розувастатин является непрофильным субстратом для метаболизма ферментами системы цитохрома Р450. Основным изоферментом, участвующим в метаболизме розувастатина, является изофермент CYP2C9. Изоферменты CYP2C19, CYP3A4 и CYP2D6 вовлечены в метаболизм в меньшей степени. Основными выявленными метаболитами розувастатина являются N-десме- тилрозувастатин и лактоновые метаболиты. N-десметилрозувастатин примерно на 50 % менее активен, чем розувастатин, лактоновые метаболиты фармакологически неактивны. Более 90 % фармакологической активности по ингибированию циркулирующей ГМГ-КоА-редуктазы обеспечивается розувастатином, остальное - его метаболитами. Выведение Около 90 % дозы розувастатина выводится в неизмененном виде через кишечник (включаяабсорбированный и неабсорбированный розувастатин). Оставшаяся часть выводится почками. Плазменный период полувыведения (Т1/2) составляет примерно 19 часов. Период полувыведения не изменяется при увеличении дозы препарата. Средний геометрический плазменный клиренс составляет приблизительно 50 л/час (коэффициент вариации 21,7 %). Как и в случае других ингибиторов ГМГ-КоА- редуктазы, в процесс "печеночного" захвата розувастатина вовлечен мембранный переносчик холестерина, выполняющий важную роль в печеночной элиминации розувастатина. Линейность Системная экспозиция розувастатина увеличивается пропорционально дозе. Фармакокинетические параметры не изменяются при ежедневном приеме. Особые популяции больных. Возраст и пол Пол и возраст не оказывают клинически значимого влияния на фармакокинетику розувастатина. Этнические группы Фармакокинетические исследования показали приблизительно двукратное увеличение медианы AUC (площади под кривой "концентрация-время") и Сmах (максимальной концентрации в плазме крови) розувастатина у пациентов монголоидной расы (японцев, китайцев, филиппинцев, вьетнамцев и корейцев) по сравнению с европеоидами: у индусов показано увеличение медианы AUC и Сmах в 1.3 раза. Фармакокинетический анализ не выявил клинически значимых различий в фармакокинетике среди европеоидов и представителей негроидной расы. Почечная недостаточность У пациентов с легкой и умеренно выраженной почечной недостаточностью величина плазменной концентрации розувастатина или N-десметилрозувастатина существенно не меняется. У пациентов с выраженной почечной недостаточностью (клиренс креатинина (КК) менее 30 мл/мин.) концентрация розувастатина в плазме крови в 3 раза выше, а концентрация N-десметилрозувастатина в 9 раз выше, чем у здоровых добровольцев. Концентрация розувастатина в плазме крови у пациентов на гемодиализе была примерно на 50 % выше, чем у здоровых добровольцев. Печеночная недостаточность У пациентов с различными стадиями печеночной недостаточности не выявлено увеличение периода полувыведения розувастатина у пациентов с 7-ю баллами и ниже по шкале Чайлд-Пью. У двух пациентов с 8-ю и 9-ю баллами по шкале Чайлд-Пью отмечено увеличение периода полувыведения, по крайней мере, в 2 раза. Опыт применения розувастатина у пациентов с более чем 9-ю баллами по шкале Чайлд-Пью отсутствует. Генетический полиморфизм Ингибиторы ГМГ-КоА-редуктазы, в том числе, розувастатин, связываются с транспортными белками ОАТР1В1 (полипептид транспорта органических анионов, участвующий в захвате статинов гепатоцитами) и BCRP (эффлюксный транспортер). У носителей генотипов SLC01B1 (ОАТР1В1) с.521СС и ABCG2 (BCRP) с.421АА отмечалось увеличение экспозиции (AUC) к розувастатину в 1,6 и 2,4 раза, соответственно, по сравнению с носителями генотипов SLC01B1 с.521ТТ и ABCG2 с.421СС. Особые указания Почечные эффекты У пациентов, получавших высокие дозы препарата Розувастатин-СЗ (в основном 40 мг), наблюдалась канальцевая протеинурия, которая в большинстве случаев была транзиторной. Такая протеинурия не свидетельствовала об остром заболевании почек или прогрессировании заболевания почек. У пациентов, принимающих препарат в дозе 40 мг, рекомендуется контролировать показатели функции почек во время лечения. Со стороны опорно-двигательного аппарата При применении препарата Розувастатин-СЗ во всех дозах и, в особенности при приеме доз препарата, превышающих 20 мг, сообщалось о следующих воздействиях на опорно-двигательный аппарат: миалгия, миопатия, в редких случаях рабдомиолиз. Определение креатинфосфокиназы Определение активности КФК не следует проводить после интенсивных физических нагрузок или при наличии других возможных причин увеличения активности КФК, что может привести к неверной интерпретации полученных результатов. В случае если исходная активность КФК существенно повышена (в 5 раз выше, чем верхняя граница нормы), через 5-7 дней следует провести повторное измерение. Не следует начинать терапию, если повторный тест подтверждает исходную активность КФК (выше более чем в 5 раз по сравнению с верхней границей нормы). До начала терапии При назначении препарата Розувастатин-СЗ, также как и при назначении других ингибиторов ГМГ-КоА-редуктазы, следует проявлять осторожность пациентам с имеющимися факторами риска миопатии/рабдомиолиза (см. раздел "С осторожностью"), необходимо рассмотреть соотношение риска и возможной пользы терапии и проводить клиническое наблюдение. Во время терапии Следует проинформировать пациента о необходимости немедленного сообщения врачу о случаях неожиданного появления мышечных болей, мышечной слабости или спазмах, особенно в сочетании с недомоганием и лихорадкой. У таких пациентов следует определять активность КФК. Терапия должна быть прекращена, если активность КФК значительно повышена (более чем в 5 раз по сравнению с верхней границей нормы) или если симптомы со стороны мышц резко'выражены и вызывают ежедневный дискомфорт (даже, если активность КФК в 5 раз меньше по сравнению с верхней границей нормы). Если симптомы исчезают, и активность КФК возвращается к норме, следует рассмотреть вопрос о повторном назначении препарата Розувастатин-СЗ или других ингибиторов ГМГ-КоА-редуктазы в меньших дозах при тщательном наблюдении за пациентом. Рутинный контроль активности КФК при отсутствии симптомов нецелесообразен. Отмечены очень редкие случаи иммуноопосредованной некротизирующей миопатии с клиническими проявлениями в виде стойкой слабости проксимальных мышц и повышения уровня КФК в сыворотке крови во время лечения или при прекращении приема статинов, в том числе розувастатина. Может потребоваться проведения дополнительных исследований мышечной и нервной системы, серологических исследований, а также терапия иммунодепрессивными средствами. Не отмечено признаков увеличения воздействия на скелетную мускулатуру при приеме препарата Розувастатин-СЗ и сопутствующей терапии. Однако сообщалось об увеличении числа случаев миозита и миопатии у пациентов, принимавших другие ингибиторы ГМГ-КоА-редуктазы в сочетании с производными фибриновой кислоты, включая гемфиброзил, циклоспорин, никотиновую кислоту, азольные противогрибковые средства, ингибиторы протеаз и макролидные антибиотики. Гемфиброзил увеличивает риск возникновения миопатии при совместном применении с некоторыми ингибиторами ГМГ-КоА-редуктазы. Таким образом, не рекомендуется одновременное применение препарата Розувастатин-СЗ и гемфиброзила. Должно быть тщательно взвешено соотношение риска и возможной пользы при совместном применении препарата Розувастатин-СЗ и фибратов или липидснижающих доз никотиновой кислоты. Противопоказан прием препарата Розувастатин-СЗ в дозе 40 мг совместно с фибратами (см. разделы "Взаимодействие с другими лекарственными препаратами" и "Противопоказания"). Через 2-4 недели после начала лечения и/или при повышении дозы препарата Розувастатин-СЗ необходим контроль показателей липидного обмена (при необходимости требуется коррекция дозы). Печень Рекомендуется проводить определение показателей функции печени до начала терапии и через 3 месяца после начала терапии. Прием препарата Розувастатин-СЗ следует прекратить или уменьшить дозу препарата, если активность трансаминаз в сыворотке крови в 3 раза превышает верхнюю границу нормы. У пациентов с гиперхолестеринемией вследствие гипотиреоза или нефротического синдрома терапия основных заболеваний должна проводиться до начала лечения препаратом Розувастатин-СЗ. Особые популяции. Этнические группы В ходе фармакокинетических исследований среди китайских и японских пациентов отмечено увеличение системной концентрации розувастатина по сравнению с показателями, полученными среди пациентов - европеоидов (см. разделы "Способ применения и дозы" и "Фармакокинетика"). Ингибиторы протеазы ВИЧ Не рекомендуется совместное применение препарата с ингибиторами протеазы ВИЧ (см. разделы "Взаимодействие с другими лекарственными препаратами" и "Противопоказания"). Лактоза Препарат не следует применять у пациентов с лактазной недостаточностью, непереносимостью галактозы и глюкозо-галактозной мальабсорбцией. Интерстициальное заболевание легких При применении некоторых статинов, особенно в течение длительного времени, сообщалось о единичных случаях интерстициального заболевания легких. Проявлениями заболевания могут являться одышка, непродуктивный кашель и ухудшение общего самочувствия (слабость, снижение веса и лихорадка). При подозрении на интерстициальное заболевание легких следует прекратить терапию статинами. Сахарный диабет 2-го типа У пациентов с концентрацией глюкозы от 5,6 до 6,9 ммоль/л терапия препаратом Розувастатин-СЗ ассоциировалась с повышенным риском развития сахарного диабета 2-го типа. Влияние на способность управлять транспортными средствами: Не проводилось исследований по изучению влияния препарата Розувастатин-СЗ на способность управлять транспортным средством и использовать механизмы. Следует соблюдать осторожность при управлении автотранспортом или работе, требующей повышенной концентрации внимания и быстроты психомоторных реакций (во время терапии может возникать головокружение). Условия хранения и отпуска из аптек Условия хранения:В сухом, защищенном от света месте, при температуре не выше 25 °С. Хранить в недоступном для детей месте. Отпуск из аптек: По рецепту Регистрационные данные Торговое название Розувастатин-СЗ Международное непатентованное название:Розувастатин. Форма выпуска:таблетки, покрытые пленочной оболочкой. Состав:Дозировка 5 мг Активное вещество: розувастатина кальция в пересчете на розувастатин - 5 мг. Вспомогательные вещества: ядро - лактозы моногидрат (сахар молочный) - 32,9 мг: кальция гидрофосфат дигидрат - 5,0 мг: повидон (поливинилпирролидон среднемолекулярный) - 3,0 мг: кроскармеллоза натрия (примеллоза) - 3,0 мг: натрия стеарилфума- рат - 0,8 мг: кремния диоксид коллоидный (аэросил) - 0,3 мг: целлюлоза микрокристаллическая - 30,0 мг: оболочка - Опадрай II (спирт поливиниловый, частично гидролизованный - 0,88 мг: макрогол (полиэтиленгликоль) 3350 - 0,247 мг: тальк - 0,4 мг: титана диоксид Е 171 - 0,3834 мг: лецитин соевый Е 322 - 0,07 мг: алюминиевый лак на основе красителя индигокармин - 0,0012 мг: алюминиевый лак на основе красителя азорубин - 0,0102 мг: алюминиевый лак на основе красителя пунцовый [Понсо 4R] - 0,0082 мг). Дозировка 10 мг Активное вещество: розувастатина кальция в пересчете на розувастатин - 10 мг. Вспомогательные вещества: ядро - лактозы моногидрат (сахар молочный) - 44,3 мг: кальция гидрофосфат дигидрат - 10,0 мг: повидон (поливинилпирролидон среднемолекулярный) - 6,0 мг: кроскармеллоза натрия (примеллоза) - 4,0 мг: натрия стеарилфума- рат - 1,2 мг: кремния диоксид коллоидный (аэросил) - 0,5 мг: целлюлоза микрокристаллическая - 44,0 мг: оболочка - Опадрай II (спирт поливиниловый, частично гидролизованный - 1,76 мг: макрогол (полиэтиленгликоль) 3350 - 0,494 мг: тальк - 0,8 мг: титана диоксид Е 171 - 0,7668 мг: лецитин соевый Е 322 - 0,14 мг: алюминиевый лак на основе красителя индигокармин - 0,0024 мг: алюминиевый лак на основе красителя азорубин - 0,0204 мг: алюминиевый лак на основе красителя пунцовый [Понсо 4R] - 0,0164 мг). Дозировка 20 мг Активное вещество: розувастатина кальция в пересчете на розувастатин - 20 мг. Вспомогательные вещества: ядро - лактозы моногидрат (сахар молочный) - 67,6 мг: кальция гидрофосфат дигидрат - 20,0 мг: повидон (поливинилпирролидон среднемолекулярный) - 9,0 мг: кроскармеллоза натрия (примеллоза) - 6,6 мг: натрия стеарилфумарат - 2,0 мг: кремния диоксид коллоидный (аэросил) - 0,8 мг: целлюлоза микрокристаллическая - 74,0 мг: оболочка - Опадрай II (спирт поливиниловый, частично гидролизованный - 2,64 мг: макрогол (полиэтиленгликоль) 3350 - 0,741 мг: тальк - 1,2 мг: титана диоксид Е 171 - 1,1502 мг: лецитин соевый Е 322 - 0,21 мг: алюминиевый лак на основе красителя индигокармин - 0,0036 мг: алюминиевый лак на основе красителя азорубин - 0,0306 мг: алюминиевый лак на основе красителя пунцовый [Понсо 4R] - 0,0246 мг). Дозировка 40 мг Активное вещество: розувастатина кальция в пересчете на розувастатин - 40 мг. Вспомогательные вещества: ядро - лактозы моногидрат (сахар молочный) - 55,2 мг: кальция гидрофосфат дигидрат - 40,0 мг: повидон (поливинилпирролидон среднемолекулярный) - 13,0 мг: кроскармеллоза натрия (примеллоза) - 8,3 мг: натрия стеарилфумарат - 2,5 мг: кремния диоксид коллоидный (аэросил) - 1,0 мг: целлюлоза микрокристаллическая - 90,0 мг: оболочка - Опадрай II (спирт поливиниловый, частично гидролизованный - 3,52 мг: макрогол (полиэтиленгликоль) 3350 - 0,988 мг: тальк - 1,6 мг: титана диоксид Е 171 - 1,5336 мг: лецитин соевый Е 322 - 0,28 мг: алюминиевый лак на основе красителя индигокармин - 0,0048 мг: алюминиевый лак на основе красителя азорубин - 0,0408 мг: алюминиевый лак на основе красителя пунцовый [Понсо 4R] - 0,0328 мг). АТХ: Регистрация: Лекарственное средство ЛП-002471 Фармгруппа: гиполипидемическое средство - ГМГ-КоА редуктазы ингибитор. Дата регистрации: 21.05.2014/19.02.2016. Окончание регстрации: 2019-05-21 2019-05-21 . Описание:Таблетки, покрытые пленочной оболочкой розового цвета, круглые, двояковыпуклые. На поперечном разрезе ядро таблетки белого или почти белого цвета. Упаковка:Таблетки, покрытые пленочной оболочкой, 5 мг, 10 мг, 20 мг и 40 мг. По 10, 14 или 30 таблеток в контурную ячейковую упаковку. По 20 или 90 таблеток в банки полимерные или во флаконы полимерные. Каждую банку или флакон, 3, 6 контурных ячейковых упаковок по 10 таблеток, 2, 4 контурных ячейковых упаковок по 14 таблеток или 2, 3, 4 контурные ячейковые упаковки по 30 таблеток вместе с инструкцией по применению помещают в картонную пачку. Срок годности:3 года. Не использовать по истечении срока годности, указанного на упаковке. Владелец рег.удостоверения:СЕВЕРНАЯ ЗВЕЗДА, ЗАО Производитель:СЕВЕРНАЯ ЗВЕЗДА, ЗАО. Представительство
730 Руб.Клопидогрел-СЗ таб. п/о 75мг №28

Показания Профилактика атеротромботических событий у пациентов, перенесших инфаркт миокарда (с давностью от нескольких дней до 35 дней), ишемический инсульт (с давностью от 7 дней до 6 месяцев) или имеющих с диагностированную окклюзионную болезнь периферических артерий. Предотвращение атеротромботических событий (в комбинации с ацетилсалициловой кислотой) у пациентов с острым коронарным синдромом: - без подъема сегмента ST (нестабильная стенокардия или инфаркт миокарда без зубца Q), включая пациентов, которым было проведено стентирование при чрескожном коронарном вмешательстве: - с подъемом сегмента ST (острый инфаркт миокарда) при медикаментозном лечении и возможности проведения тромболизиса. Противопоказания Противопоказания - Повышенная чувствительность к клопидогрелу или любому из вспомогательных веществ препарата: - тяжелая печеночная недостаточность: - острое кровотечение, например, кровотечение из пептической язвы или внутричерепное кровоизлияние: - редкая наследственная непереносимость лактозы, дефицит лактазы и глюкозо-галактозная мальабсорбция: - беременность и период лактации (см. "Беременность и период грудного вскармливания"): - детский возраст до 18 лет (безопасность и эффективность применения не установлены). С осторожностью: - Умеренная печеночная недостаточность, при которой возможна предрасположенность к кровотечению (ограниченный клинический опыт применения): - почечная недостаточность (ограниченный клинический опыт применения): - травма, хирургические вмешательства (см. "Особые указания"): - заболевания, при которых имеется предрасположенность к развитию кровотечений (особенно желудочно-кишечных или внутриглазных): - одновременный прием нестероидных противовоспалительных препаратов, в том числе и селективных ингибиторов циклооксигеназы-2 (ЦОГ-2): - одновременное назначение варфарина, гепарина, ингибиторов гликопротеина IIb/IIIа: - у пациентов с генетически обусловленным снижением функции изофермента CYP2C19 в рекомендованных дозах (имеются литературные данные, указывающие на то, что пациенты с генетически обусловленным снижением функции изофермента СYP2C19 подвергаются меньшей системной экспозиции активным метаболитом клопидогрела и имеют менее выраженное действие препарата, кроме этого у них может наблюдаться большая частота сердечно-сосудистых осложнений после инфаркта миокарда по сравнению с пациентами с нормальной функцией изофермента CYP2C19). Беременность В качестве меры предосторожности не рекомендуется прием клопидогрела во время беременности из-за отсутствия клинических данных по его приему беременными женщинами, хотя исследования на животных и не выявили ни прямых, ни непрямых неблагоприятных эффектов на течение беременности, эмбриональное развитие, роды и постнатальное развитие. Кормление грудью в случае лечения клопидогрелом следует прекратить, так как в исследованиях на крысах было показано, что клопидогрел и/или его метаболиты экскретируются в грудное молоко. Проникает ли клопидогрел в грудное молоко человека - неизвестно. Применение и дозы Взрослые и пациенты пожилого возраста с нормальной активностью изофермента CYP2C19 Клопидогрел следует принимать внутрь, независимо от приема пищи. Инфаркт миокарда, ишемический инсульт и диагностированная окклюзионная болезнь периферических артерий Препарат принимается по 75 мг один раз в сутки. У больных с инфарктом миокарда (ИМ) лечение можно начинать с первых дней до 35 дня ИМ, а у больных с ишемическим инсультом (ИИ) - в сроки от 7 дней до 6 месяцев после ИИ. Острый коронарный синдром без подъема сегмента ST (нестабильная стенокардия, инфаркт миокарда без зубца Q) Лечение препаратом Клопидогрел должно быть начато с однократного приема нагрузочной дозы, составляющей 300 мг, а затем продолжено приемом дозы 75 мг один раз в сутки (в сочетании с ацетилсалициловой кислотой в качестве антиагрегантного средства в дозах 75-325 мг в сутки). Поскольку применение более высоких доз ацетилсалициловой кислоты связано с увеличением риска кровотечений, рекомендуемая при этом показании доза ацетилсалициловой кислоты не должна превышать 100 мг. Максимальный терапевтический эффект наблюдается к третьему месяцу лечения. Курс лечения до 1 года. Острый коронарный синдром с подъемом сегмента ST (острый инфаркт миокарда с подъемом сегмента ST) Клопидогрел назначается однократно в дозе 75 мг один раз в день с первоначальным однократным приемом нагрузочной дозы в комбинации с ацетилсалициловой кислотой в качестве антиагрегантного средства и тромболитиками (или без тромболитиков). Комбинированную терапию начинают как можно раньше после появления симптомов и продолжают в течение, по крайней мере, четырех недель. У пациентов старше 75 лет лечение препаратом Клопидогрел должно начинаться без приема его нагрузочной дозы. Пациенты с генетически обусловленным снижением функции изофермента CYP2C19 Ослабление метаболизма с помощью изофермента CYP2C19 может приводить к уменьшению антиагрегантного действия клопидогрела. Оптимальный режим дозирования для пациентов с ослаблением метаболизмом с помощью изофермента CYP2C19 пока не установлен. Пациенты с нарушением функции почек После повторных приемов препарата Клопидогрел в дозе 75 мг/сутки у пациентов с тяжелым поражением почек (клиренс креатинина от 5 до 15 мл/мин) ингибирование АДФ-индуцированной агрегации тромбоцитов (25 %) было ниже по сравнению с таковым у здоровых добровольцев, однако, удлинение времени кровотечения было подобным таковому у здоровых добровольцев, получавших Клопидогрел в дозе 75 мг в сутки. Кроме этого, у всех пациентов была хорошая переносимость препарата. Пациенты с нарушением функции печени После ежедневного в течение 10 дней приема Клопидогрела в дозе 75 мг у больных с тяжелым поражением печени ингибирование АДФ-индуцированной агрегации тромбоцитов было подобным таковому у здоровых добровольцев. Среднее время кровотечения было также сопоставимо в обеих группах. Пациенты различной этнической принадлежности Распространенность аллелей генов изофермента CYP2C19, отвечающих за промежуточный и сниженный метаболизм клопидогрела до его активного метаболита, различается у представителей различных этнических групп (см. раздел "Фармакогенетика"). Имеются лишь ограниченные данные для представителей монголоидной расы по оценке влияния генотипа изофермента CYP2C19 на клинические результирующие события. Побочные эффекты и передозировка Побочные эффекты: Классификация частоты развития побочных эффектов (ВОЗ): часто - более 1/100 и менее 1/10, нечасто - более 1/1000 и менее 1/100, редко - более 1/10000 и менее 1/1000, очень редко - менее 1/10000). Со стороны центральной и периферической нервной системы: нечасто - головная боль, головокружение и парестезии: редко - вертиго: очень редко - нарушение вкусовых ощущений. Со стороны пищеварительной системы: часто - диарея, абдоминальные боли, диспепсия: нечасто - язва желудка и двенадцатиперстной кишки, гастрит, рвота, тошнота, запор, метеоризм: очень редко - панкреатит, колит (в том числе язвенный или лимфоцитарный колит), стоматит. Нарушения со стороны гемостаза: нечасто - удлинение времени кровотечения. Нарушения со стороны кроветворения: нечасто - тромбоцитопения, лейкопения, нейтропения и эозинофилия, очень редко - тромбоцитопеническая тромбогемолитическая пурпура, тяжелая тромбоцитопения (число тромбоцитов <: 30x 109/л), агранулоцитоз, гранулоцитопения, апластическая анемия (панцитопения), анемия. Нарушения со стороны кожи и подкожных тканей: нечасто - кожная сыпь и зуд: очень редко - ангионевротический отек, крапивница, эритематозная сыпь (связанные с клопидогрелом или ацетилсалициловой кислотой): очень редко - буллезный дерматит (многоформная эритема, синдром Стивенса-Джонсона, токсический эпидермальный некролиз), экзема и плоский лишай. Нарушения со стороны имунной системы: очень редко - анафилактоидные реакции, сывороточная болезнь. Психические расстройства: очень редко - спутанность сознания, галлюцинации. Нарушения со стороны сосудистой системы: очень редко - васкулит, выраженное снижение артериального давления (АД), внутричерепное кровоизлияние, глазные кровоизлияния (конъюнктивальные, в ткани и сетчатку глаза), гематома, носовое кровотечение, кровотечение из дыхательных путей, желудочно-кишечное кровотечение, забрюшинное кровоизлияние со смертельным исходом, кровоизлияния в мышцы и суставы, гематурия и др. Нарушения со стороны органов дыхания: очень редко - бронхоспазм, интерстициальный пневмонит. Нарушения со стороны гепато-бинарной системы: очень редко - острая печеночная недостаточность, гепатит. Нарушения со стороны опорно-двигательного аппарата: очень редко - артралгия, артрит, миалгия. Нарушения со стороны почек и мочевыводящих путей: очень редко - гломерулонефрит. Общие нарушения: очень редко - лихорадка. Изменения лабораторных показателей: очень редко - изменение печёночных проб, увеличение концентрации сывороточного креатинина. Передозировка: Симптомы: удлинение времени кровотечения с последующими осложнениями в виде развития кровотечений. Лечение: остановка кровотечения, переливание тромбоцитарной массы. Антидот неизвестен. Взаимодействие с другими ЛС: Варфарин: одновременный прием вместе с клопидогрелом может увеличить интенсивность кровотечений, поэтому применение этой комбинации не рекомендуется. Блокаторы IIb/IIIa-рецепторов: назначение блокаторов IIb/IIIа-рецепторов совместно с клопидогрелом, требует осторожности у пациентов, имеющих повышенный риск развития кровотечения (при травмах и хирургических вмешательствах или других патологических состояниях) (см. раздел "Особые указания"). Ацетилсалициловая кислота: ацетилсалициловая кислота не изменяет эффекта клопидогрела, ингибирующего АДФ-индуцируемую агрегацию тромбоцитов, но клопидогрел потенцирует влияние ацетилсалициловой кислоты на коллаген-индуцируемую агрегацию тромбоцитов. Тем не менее, одновременный с клопидогрелом прием ацетилсалициловой кислоты, в качестве жаропонижающего средства, по 500 мг 2 раза в сутки в течение 1 дня не вызывал существенного увеличения времени кровотечения, вызываемого приемом клопидогрела. Между клопидогрелом и ацетилсалициловой кислотой возможно фармакодинамическое взаимодействие, которое приводит к повышению риска кровотечения. Поэтому при их одновременном применении следует соблюдать осторожность, хотя в клинических исследованиях пациенты получали комбинированную терапию клопидогрелом и ацетилсалициловой кислотой до одного года. Гепарин: по данным клинического исследования, проведенного с участием здоровых добровольцев, при приеме клопидогрела не требовалось изменения дозы гепарина и не изменялось его антикоагулянтное действие. Одновременное применение гепарина не изменяло антиагрегантного эффекта клопидогрела. Между Клопидогрелом и гепарином возможно фармакодинамическое взаимодействие, которое может увеличивать риск развития кровотечений, поэтому одновременное применение этих препаратов требует осторожности. Тромболитики: безопасность одновременного применения клопидогрела, фибринспецифических или фибриннеспецифических тромболитических препаратов и гепарина была исследована у больных с острым инфарктом миокарда. Частота клинически значимых кровотечений была аналогична той, которая наблюдалась в случае одновременного применения тромболитических средств и гепарина с ацетилсалициловой кислотой. Нестероидные противовоспалительные препараты (НПВП): в клиническом исследовании, проведенным с участием здоровых добровольцев, одновременное применение клопидогрела и напроксена увеличивало скрытые потери крови через ЖКТ. Однако в связи с отсутствием исследований по взаимодействию клопидогрела с другими НПВП, в настоящее время не известно, имеется ли повышенный риск желудочно-кишечных кровотечений при приеме клопидогрела вместе с другими НПВП. Поэтому назначение НПВП, в том числе селективных ингибиторов ЦОГ-2, в сочетании с клопидогрелом следует проводить с осторожностью (см. раздел "Особые указания"). Другая комбинированная терапия Так как клопидогрел метаболизируется до образования своего активного метаболита частично при помощи изофермента CYP2C19, использование препаратов ингибирующих эту систему может привести к снижению уровня активного метаболита клопидогрела и уменьшению его клинической эффективности. Одновременный прием препаратов, ингибирующих изофермент CYP2C19 (например, омепразол) не рекомендуется. Был проведен ряд клинических исследований с клопидогрелом и другими одновременно назначаемыми препаратами с целью изучения возможных фармакодинамических и фармакокинетических взаимодействий, которые показали, что: - при применении клопидогрела совместно с атенололом, нифедипином или с обоими препаратами одновременно клинически значимого фармакодинамического взаимодействия не наблюдалось: - одновременное применение фенобарбитала, циметидина и эстрогенов не оказало существенного влияния на фармакодинамику клопидогрела: - фармакокинетические показатели дигоксина и теофиллина не изменялись при их одновременном применении с клопидогрелом. - антацидные средства не уменьшали абсорбцию клопидогрела. - фенитоин и толбутамид можно с безопасностью применять одновременно с клопидогрелом, несмотря на то, что данные, полученные в ходе исследований с микросомами печени человека, свидетельствуют о том, что карбоксильный метаболит клопидогрела может ингибировать активность изофермента CYP2C9, что может приводить к повышению плазменных концентраций некоторых лекарственных средств (фенитоина, толбутамида и некоторых НПВП), которые метаболизируются с помощью изофермента CYP2C9. - ингибиторы ангиотензин-превращающего фермента (АПФ), диуретические средства, бета-адреноблокаторы, блокаторы "медленных" кальциевых каналов, гипогликемические средства (в т.ч. инсулин), гиполипидемические средства, противоэпилептические средства, гормонозаместительная терапия и блокаторы GPIIb/IIIa-рецепторов: в клинических исследованиях не было выявлено клинически значимых нежелательных взаимодействий. Фармакологическое действие и фармакокинетика Клопидогрел представляет собой пролекарство, один из активных метаболитов которого является ингибитором агрегации тромбоцитов. Активный метаболит клопидогрела селективно ингибирует связывание аденозиндифосфата (АДФ) с P2Y12 рецептором тромбоцитов и последующую АДФ-опосредованную активацию комплекса GPIIb/IIIa, приводя к подавлению агрегации тромбоцитов. Благодаря необратимому связыванию тромбоциты остаются невосприимчивыми к стимуляции АДФ в течение всего оставшегося срока своей жизни (примерно 7-10 дней), а восстановление нормальной функции тромбоцитов происходит со скоростью, соответствующей скорости обновления тромбоцитов. Агрегация тромбоцитов, вызванная агонистами, отличными от АДФ, также ингибируется за счет блокады усиленной активации тромбоцитов высвобождаемым АДФ. Так как образование активного метаболита происходит при помощи изоферментов системы Р450, некоторые из которых могут отличаться полиморфизмом или могут ингибироваться другими препаратами, не у всех пациентов возможно адекватное подавление тромбоцитов. Клопидогрел способен предотвращать развитие атеротромбоза при любых локализациях атеросклеротического поражения сосудов, в частности при поражениях церебральных, коронарных или периферических артерий. При ежедневном приеме клопидогрела в дозе 75 мг с первого же дня приема отмечается значительное подавление АДФ-индуцируемой агрегации тромбоцитов, которое постепенно увеличивается в течение 3-7 дней и затем выходит на постоянный уровень (при достижении равновесного состояния). В равновесном состоянии агрегация тромбоцитов подавляется в среднем на 40-60%. После прекращения приема клопидогрела агрегация тромбоцитов и время кровотечения постепенно возвращаются к исходному уровню в среднем в течение 5 дней. Фармакокинетика: Всасывание При курсовом приеме внутрь в дозе 75 мг в сутки клопидогрел быстро всасывается. Средние максимальные концентрации неизмененного клопидогрела в плазме крови (примерно 2,2-2,5 нг/мл после приема внутрь разовой дозы 75 мг) достигаются примерно через 45 минут после приема. По данным экскреции метаболитов клопидогрела почками его абсорбция составляет примерно 50%. Распределение In vitro клопидогрел и его основной циркулирующий в крови неактивный метаболит обратимо связываются с белками плазмы крови (на 98% и 94%, соответственно) и данная связь является ненасыщаемой вплоть до концентрации 100 мг/л. Метаболизм Клопидогрел интенсивно метаболизируется в печени. In vitro и in vivo клопидогрел метаболизируется двумя путями: первый - через ферменты и последующий гидролиз с образованием неактивного производного карбоксильной кислоты (85% от циркулирующих метаболитов), а второй путь - через систему цитохрома Р450. Вначале клопидогрел метаболизируется до 2-оксо-клопидогрела, являющегося промежуточным метаболитом. Последующий метаболизм 2-оксо-клопидогрела приводит к образованию активного метаболита клопидогрела - тиольного производного клопидогрела. In vitro этот путь метаболизма происходит при помощи изоферментов CYP2CI9, CYP1A2 и CYP2B6. Активный тиольный метаболит клопидогрела, который был выделен в исследованиях in vitro, быстро и необратимо связывается с рецепторами тромбоцитов, блокируя, таким образом, агрегацию тромбоцитов. Выведение В течение 120 часов после приёма внутрь человеком 14С-меченого клопидогрела около 50% радиоактивности выделяется почками и приблизительно 46% радиоактивности - через кишечник. После однократного приема внутрь дозы в 75 мг период полувыведения клопидогрела составляет примерно 6 часов. После однократного приема и приема повторных доз период полувыведения основного циркулирующего в крови неактивного метаболита составляет 8 часов. Фармакогенетика С помощью изофермента CYP2C19 образуются как активный метаболит, так и промежуточный метаболит - 2-оксо-клопидогрел. Фармакокинетика и антиагрегантное действие активного метаболита клопидогрела, при исследовании агрегации тромбоцитов ex vivo, варьируют в зависимости от генотипа изофермента CYP2C19. Аллель гена СУР2С191 соответствует полностью функциональному метаболизму, тогда как аллели генов CYP2C192 и CYP2C193 являются нефункциональными. Аллели генов СУР2С192 и СУР2С193 являются причиной снижения метаболизма у большинства представителей европеоидной (85%) и монголоидной расы (99%). Другие аллели, с которыми связывается отсутствие или снижение метаболизма, встречаются реже и включают, но не ограничиваются аллелями генов CYP2C19M, 5, 6, 7 и *8. Пациенты с низкой активностью изофермента CYP2C19, должны обладать двумя указанными выше аллелями гена с потерей функции. Опубликованные частоты встречаемости фенотипов лиц с низкой активностью изофермента СYP2C19 составляют у лиц европеоидной расы 2%, у лиц негроидной расы 4% и у китайцев 14%. Для определения имеющегося у пациента генотипа изофермента CYP2C19 существуют соответствующие тесты. По данным перекрестного исследования (40 добровольцев) и по данным мета-анализа шести исследований (335 добровольцев). В которые входили лица с очень высокой, высокой, промежуточной и низкой активностью изофермента CYP2C19, каких-либо существенных различий в экспозиции активного метаболита и в средних значениях ингибирования агрегации тромбоцитов (ИАТ) (индуцированной АДФ) у добровольцев с очень высокой, высокой и промежуточной активностью изофермента CYP2C19 выявлено не было. У добровольцев с низкой активностью изофермента CYP2C19 экспозиция активного метаболита снижалась по сравнению с добровольцами с высокой активностью изофермента CYP2C19. Когда добровольцы с низкой активностью изофермента CYP2C19 получали схему лечения 600 мг нагрузочная доза/150 мг поддерживающая доза (600 мг/150 мг), экспозиция активного метаболита была выше, чем при приеме схемы лечения 300 мг/75 мг. Кроме этого, ИАТ было подобно таковому в группах пациентов с более высокой интенсивностью метаболизма с помощью изофермента CYP2C19, получавших схему лечения 300 мг/75 мг. Однако в исследованиях с учетом клинических исходов режим дозирования клопидогрела для пациентов этой группы (пациентов с низкой активностью изофермента CYP2C19) пока не установлен. Проведенные до настоящего времени клинические исследования не имели достаточного объема выборки для выявления различий в клиническом исходе у пациентов с низкой активностью изофермента CYP2C19. Отдельные группы пациентов Фармакокинетика активного метаболита клопидогрела у пациентов пожилого возраста, детей, пациентов с заболеваниями почек и печени не изучена. Особые указания При лечении препаратом Клопидогрел, особенно в течение первых недель лечения и/или после инвазивных кардиологических процедур/хирургического вмешательства, необходимо вести тщательное наблюдение за пациентами на предмет исключения признаков кровотечения, в том числе и скрытого. В связи с риском развития кровотечения и гематологических нежелательных эффектов (см. раздел "Побочное действие") в случае появления в ходе лечения клинических симптомов, подозрительных на возникновение кровотечения, следует срочно сделать клинический анализ крови, определить активированное частичное тромбопластиновое время (АЧТВ), количество тромбоцитов, показатели функциональной активности тромбоцитов и провести другие необходимые исследования. Клопидогрел, также как и другие антитромбоцитарные препараты, следует применять с осторожностью у больных, имеющих повышенный риск развития кровотечения, связанный с травмами, хирургическими вмешательствами или другими патологическими состояниями, а также у больных получающих ацетилсалициловую кислоту, нестероидные противовоспалительные препараты, в том числе ингибиторы ЦОГ-2, гепарин или ингибиторы гликопротеина IIb/IIIа. Совместное применение клопидогрела с варфарином, может усилить интенсивность кровотечений (см. "Взаимодействие с другими лекарственными препаратами"), поэтому, за исключением особых редких клинических ситуаций (таких как наличие флотирующего тромба в левом желудочке, стентирование у пациентов с мерцательной аритмией) совместное применение Клопидогрела и варфарина не рекомендуется. Если больному предстоит плановое хирургическое вмешательство, и при этом нет необходимости в антитромбоцитарном эффекте, то за 7 дней до операции прием препарата Клопидогрел следует прекратить. Клопидогрел удлиняет время кровотечения и должен применяться с осторожностью у больных с заболеваниями, предрасполагающими к развитию кровотечений (особенно, желудочно-кишечных и внутриглазных). Больные должны быть предупреждены о том, что при приёме препарата Клопидогрел (одного или в комбинации с ацетилсалициловой кислотой) для остановки кровотечения может потребоваться больше времени, а также о том, что в случае возникновении у них необычного (по локализации или продолжительности) кровотечения им следует сообщить об этом своему лечащему врачу. Перед любой предстоящей операцией и перед началом приема любого нового лекарственного препарата больные должны сообщать врачу (включая стоматолога) о приеме препарата Клопидогрел. Очень редко, после применения препарата Клопидогрел (иногда даже непродолжительного) отмечались случаи развития тромбоцитопенической тромбогемолитической пурпуры (ТТП), которая характеризуется тромбоцитопенией и микроангиопатической гемолитической анемией, сопровождающимися неврологическими расстройствами, нарушением функции почек и лихорадкой. ТТП является потенциально угрожающим жизни состоянием, требующим немедленного лечения, включая плазмаферез. В период лечения необходимо контролировать функциональную активность печени. При тяжелых нарушениях функции печени следует помнить о риске развития геморрагического диатеза. Прием препарата Клопидогрел не рекомендуется при остром инсульте с давностью менее 7 дней (так как отсутствуют данные по его применению при этом состоянии). Клопидогрел не следует принимать больным с редкой наследственной непереносимостью галактозы, дефицитом лактазы и синдромом мальабсорбции глюкозы-галактозы (см. раздел "Состав"). Влияние на способность управлять транспортными средствами: Клопидогрел не оказывает существенного влияния на способность, необходимую для управления автотранспортом или работы с механизмами. Условия хранения и отпуска из аптек Условия хранения:В сухом, защищенном от света месте, при температуре не выше 25 °С. Хранить в недоступном для детей месте. Отпуск из аптек: По рецепту Регистрационные данные Торговое название Клопидогрел-СЗ Международное непатентованное название:Клопидогрел. Форма выпуска:таблетки, покрытые пленочной оболочкой. Состав:1 таблетка, покрытая пленочной оболочкой, содержит: активное вещество: клопидогрела гидросульфат в пересчете на клопидогрел или клопидогреля бисульфат в пересчете на клопидогрел 75 мг. вспомогательные вещества (ядро): лактозы моногидрат (сахар молочный) 38,4 мг, целлюлоза микрокристаллическая 129,48 мг, кроскармеллоза натрия (примеллоза) 12,0 мг, кремния диоксид коллоидный (аэросил) 3,12 мг, натрия стеарилфумарат 2,0 мг. вспомогательные вещества (оболочка): Опадрай II 8 мг (спирт поливиниловый, частично гидролизованный 3,52 мг, тальк 1,6 мг, титана диоксид Е 171 1,5336 мг, макрогол (полиэтиленгликоль 3350) 0,988 мг, лецитин соевый Е 322 0,28 мг, алюминиевый лак на основе красителя азорубин 0,0408 мг, алюминиевый лак на основе красителя пунцовый [Понсо 4R] 0,0328 мг, алюминиевый лак на основе красителя индигокармин 0,0048 мг). АТХ: Регистрация: ЛП-001884 Фармгруппа: антиагрегантное средство. Дата регистрации: 24.10.2012. Окончание регстрации: . Описание:Круглые, двояковыпуклые таблетки, покрытые пленочной оболочкой от розового до темно-розового цвета. Упаковка:Таблетки, покрытые пленочной оболочкой, 75 мг. По 10, 14 или 30 таблеток в контурной ячейковой упаковке. По 30 таблеток в банке полимерной или во флаконе полимерном. 3, 6 контурных ячейковых упаковок по 10 таблеток, 1, 2, 4 контурные ячейковые упаковки по 14 таблеток, 1, 2, 3 контурные ячейковые упаковки по 30 таблеток, банка или флакон вместе с инструкцией по применению в картонной пачке. Срок годности:3 года. Не использовать по истечении срока годности, указанного на упаковке. Владелец рег.удостоверения:СЕВЕРНАЯ ЗВЕЗДА, ЗАО Производитель:СЕВЕРНАЯ ЗВЕЗДА, ЗАО. Представительство:СЕВЕРНАЯ ЗВЕЗДА ЗАО
388 Руб.Эсмия таб. 5мг №28
Показания Предоперационная терапия симптомов миомы матки средней тяжести и тяжелой степени у взрослых женщин репродуктивного возраста старше 18 лет. Длительность терапии - не более 2 курсов. Противопоказания Противопоказания - Гиперчувствительность к улипристалу или любому из вспомогательных веществ. - Беременность и период грудного вскармливания. - Кровотечение из влагалища неясной этиологии или по причинам, не связанным с миомой матки. - Рак матки, шейки матки, яичников или молочной железы. - Продолжительность терапии более 2 курсов. - Возраст <: 18 лет. - Бронхиальная астма, тяжелая форма, не поддающаяся коррекции пероральными глюкокортикостероидами. С осторожностью: Почечная и/или печеночная недостаточность: бронхиальная астма Беременность Беременность Улипристал противопоказан при беременности. Данные о применении улипристала у беременных отсутствуют или ограничены. Несмотря на то, что в ходе исследований на животных тератогенный потенциал не выявлен, данных в отношении репродуктивной токсичности недостаточно. Период грудного вскармливания В исследованиях на животных показано, что улипристал проникает в грудное молоко. Улипристал проникает в женское грудное молоко. Воздействие препарата на новорожденных и детей младшего возраста не изучалось, поэтому нельзя исключить риск для детей в период грудного вскармливания. Улипристал противопоказан в период грудного вскармливания. Применение и дозы Внутрь по одной таблетке 1 раз в день независимо от приема пищи в течение не более 3 месяцев. Допускается однократное повторное проведение 3-месячного курса терапии. Повторный курс терапии надо начинать не ранее второго менструального цикла после окончания первого курса, в течение первой недели. Отсутствуют данные по безопасности длительных курсов терапии, поэтому длительность лечения не должна превышать двух курсов продолжительностью по 3 месяца. В случае пропуска таблетки следует принять таблетку препарата Эсмия® как можно быстрее. Если прием пропущен более чем на 12 часов, то пропущенная таблетка не принимается, и следует просто возобновить обычный режим приема. Особые группы пациенток Почечная недостаточность У пациенток с легкой или умеренной степенью почечной недостаточности коррекции дозы не требуется. Препарат Эсмия® не рекомендован для применения у пациенток с тяжелой почечной недостаточностью при невозможности постоянного наблюдения (см. раздел "Особые указания"). Печеночная недостаточность У пациенток с легкой степенью печеночной недостаточности коррекции дозы не требуется. Препарат Эсмия® не рекомендован для применения у пациенток с умеренной или тяжелой печеночной недостаточностью при невозможности постоянного наблюдения (см. раздел "Особые указания"). Дети Применение препарата Эсмия® по соответствующим показаниям у детей не предусмотрено. Безопасность и эффективность улипристала установлены только для женщин от 18 лет и старше. Побочные эффекты и передозировка Побочные эффекты: Обзор профиля безопасности Безопасность улипристала оценивалась у 602 женщин с миомами матки, получавших 5 мг или 10 мг улипристала в ходе исследований фазы 3. Наиболее частым наблюдаемым явлением в клинических исследованиях была аменорея (80,8 %), которая считается желаемым исходом. Наиболее частой побочной реакцией было появление "приливов". Подавляющее большинство побочных реакций были легкими или умеренными (93,6 %), не приводили к прекращению лечения препаратом (99,5 %) и разрешались самостоятельно. Безопасность двух курсов лечения улипристалом 10 мг, каждый продолжительностью 3 месяца, оценивалась в исследовании фазы 3 с участием женщин с миомами матки. Полученные данные сопоставимы с профилем безопасности при проведении лечения в рамках одного курса. Перечень побочных реакций В ходе 3 исследований фазы 3 у пациенток с миомами матки, получавших препарат в течение 3 месяцев, сообщалось о следующих побочных реакциях. Неблагоприятные побочные реакции представлены по системно-органным классам в соответствии с классификацией MedDRA и с частотой возникновения: очень часто (>: 1/10): часто (от >: 1/100 до <: 1/10): нечасто (от >: 1/1000 до <: 1/100): редко (от >: 1/10 000 до <: 1/1000): очень редко (<: 1/10 000): неизвестно (невозможно оценить на основании имеющихся данных). В пределах каждой частотной группы побочные реакции представлены в порядке убывания серьезности. Нарушения психики Нечастые: беспокойство, эмоциональные расстройства. Нарушения со стороны нервной системы Частые: головная боль (- см. раздел "Описание отдельных побочных реакций"). Нечастые: головокружение. Нарушения со стороны обмена веществ и питания Нечастые: увеличение массы тела. Нарушения со стороны органа слуха и лабиринтные нарушения. Частые: вертиго. Нарушения со стороны дыхательной системы, органов грудной клетки и средостения Нечастые: носовое кровотечение. Нарушения со стороны желудочно-кишечного тракта Частые: боли в животе, тошнота. Нечастые: диспепсия, сухость во рту, метеоризм, запор. Нарушения со стороны кожи и подкожных тканей. Частые: акне, повышенная потливость. Нечастые: алопеция, сухость кожи. Нарушения со стороны скелетно-мышечной системы и соединительной ткани Частые: боли в костях и мышцах. Нечастые: боли в спине. Нарушения со стороны почек и мочевыводящих путей. Нечастые: недержание мочи. Нарушения со стороны половых органов и молочной железы Очень частые: аменорея, утолщение эндометрия. Частые: "приливы", тазовая боль, киста яичника, напряженность или болезненность молочных желез. Нечастые: метроррагия, разрыв кисты яичника, выделения из влагалища, увеличение и неприятные ощущения в области молочных желез. Общие расстройства и нарушения в месте введения. Частые: отеки, повышенная утомляемость. Нечастые: астения. Изменения лабораторных и инструментальных исследований. Частые: повышение концентрации холестерина в крови. Нечастые: повышение концентрации триглицеридов в крови. Описание отдельных побочных реакций. Утолщение эндометрия У 10-15 % пациенток, получавших улипристал, может происходить утолщение эндометрия (>: 16 мм по данным УЗИ или МРТ на момент окончания лечения). Это явление обратимо после прекращения лечения и возобновления менструального цикла. Кроме того, обратимые изменения в эндометрии, обозначаемые как РАЕС, отличаются от гиперплазии эндометрия. Патоморфолог должен быть информирован о приеме пациенткой улипристала при проведении гистологического исследования при гистерэктомии или биопсии эндометрия. "Приливы" "Приливы" отмечались у 9,8% пациенток, но их частота варьировала в разных исследованиях. В исследовании с активным контролем их частота составила 24 % (10,5 % средней или тяжелой степени тяжести) для группы улипристала и 60,4 % (39,6 % средней или тяжелой степени тяжести) для группы лейпрорелина. В плацебо-контролируемом исследовании частота "приливов" составила 1,0 % для улипристала и 0 % для плацебо. В рамках открытого исследования фазы 3 частота "приливов" составила 4,3 % в группе улипристала. Головная боль Головная боль легкой или умеренной степени отмечалась у 6,8 % пациенток. *Киста яичника У 1,2 % пациенток в ходе лечения были обнаружены функциональные кисты яичника, которые спонтанно исчезли в течение нескольких недель. Маточное кровотечение Пациентки с тяжелым менструальным кровотечением, обусловленным лейомиомой матки, находятся в группе риска повышенной кровопотери, что может потребовать хирургического вмешательства. Было несколько таких сообщений, как в ходе терапии, так и через 2-3 месяца после окончания лечения улипристалом. Передозировка: Данные о передозировке улипристала ограничены. Однократные дозы до 200 мг и ежедневные дозы 50 мг на протяжении 10 дней назначались ограниченному числу добровольцев, при этом не отмечено тяжелых или серьезных нежелательных реакций. Взаимодействие с другими ЛС: Возможное влияние других лекарственных препаратов на действие улипристала Гормональные контрацептивы Улипристал обладает стероидной структурой и действует как селективный модулятор рецепторов прогестерона с преобладающим ингибирующим эффектом в отношении рецепторов прогестерона. Таким образом, гормональные контрацептивы и гестагены могут снижать эффективность улипристала путем конкурентного воздействия на рецептор прогестерона. Поэтому не рекомендуется одновременное применение препаратов, содержащих гестагены. Ингибиторы изофермента CYP3A4 После применения ингибитора средней мощности изофермента CYP3A4 эритромицина пропионата (500 мг 2 раза в день в течение 9 дней) у здоровых женщин-добровольцев показатели Cmax и AUC улипристала повышались в 1,2 и 2,9 раза соответственно: величина AUC активного метаболита улипристала повышалась в 1,5 раза, в то время как Сmах активного метаболита снижалась (в 0,52 раза). На фоне применения кетоконазола (400 мг 1 раз в сутки, 7 дней), мощного ингибитора CYP3A4, у здоровых женщин-добровольцев исследования отмечалось увеличение показателей Сmах и AUC улипристала в 2 и 5,9 раз соответственно. Отмечалось увеличение показателей AUC для активного метаболита улипристала в 2,4 раза при снижении его Сmах (изменение в 0,53 раза). Коррекции дозы при применении улипристала у пациенток, получающих слабые ингибиторы изофермента CYP3A4, не требуется. Совместное применение ингибиторов средней мощности или мощных ингибиторов изофермента CYP3A4 с улипристалом не рекомендуется. Индукторы изофермента CYP3A4 На фоне применения мощного индуктора CYP3A4 рифампицина (300 мг 2 раза в сутки, 9 дней) у здоровых женщин-добровольцев отмечалось выраженное снижение показателей Сmах и AUC улипристала и его активного метаболита более чем на 90 %. Также отмечено снижение периода полувыведения улипристала в 2,2 раза, что соответствует уменьшению его экспозиции примерно в 10 раз. Сопутствующее применение улипристала и мощных индукторов CYP3A4 (например, рифампицина, рифабутина, карбамазепина, окскарбазепина, фенитоина, фосфенитоина, фенобарбитала, примидона, зверобоя продырявленного, эфавиренза, невирапина, ритонавира - на фоне долгосрочного применения) не рекомендуется. Препараты, влияющие на pH желудочного сока Применение улипристала (10 мг/день) вместе с ингибитором протонной помпы эзомепразолом (20 мг 1 раз в день в течение 6 дней) приводит к снижению средней Сmах на 65 %, удлинению tmax (от медианы 0,75 часа до 1,0 часа) и повышению средней AUC на 13 %. Такое действие лекарственных препаратов, повышающих pH желудочного сока, не считается клинически значимым для ежедневного приема таблеток улипристала. Возможное влияние улипристала на действие других лекарственных препаратов Гормональные контрацептивы Улипристал может препятствовать действию гормональных контрацептивов (только гестагенсодержащих таблеток, гестагенвысвобождающих систем или комбинированных пероральных контрацептивов) и препаратов гестагена, применяемых по иным показаниям. Поэтому сопутствующее применение лекарственных препаратов, содержащих гестаген, не рекомендовано. Гестагенсодержащие препараты не следует применять в течение 12 дней после прекращения лечения улипристалом. Субстраты Р-гликопротеина Данные in vitro показывают, что в клинически значимых концентрациях улипристал в процессе всасывания в стенке желудочно-кишечного тракта может являться ингибитором Р-гликопротеина (P-gp). Одновременное применение улипристала и субстрата P-gp не исследовалось, поэтому нельзя исключить вероятность взаимодействий. Данные in vivo свидетельствуют о том, что прием улипристала (таблетка 10 мг) за 1,5 часа до приема субстрата P-gp фексофенадина (60 мг) не оказывает клинически значимого воздействия на фармакокинетику фексофенадина. Таким образом, рекомендуется соблюдать интервал не менее 1,5 часов между приемом улипристала и субстратов P-gp (например, дабигатрана этексилата, дигоксина, фексофенадина). Пациентка должна сообщить лечащему врачу о всех лекарственных препаратах, которые она принимает, даже если они отпускаются без рецепта. Фармакологическое действие и фармакокинетика Улипристал - активный при приеме внутрь синтетический селективный модулятор рецепторов прогестерона (СМПР), характеризующийся тканеспецифичным частичным антипрогестероновым эффектом. Эндометрий Улипристал оказывает прямое действие на эндометрий. При начале ежедневного приема препарата в дозе 5 мг в течение менструального цикла у большинства женщин (включая пациенток с миомой) заканчивается очередное менструальное кровотечение, а следующее не наступает. Когда прием препарата прекращается, менструальный цикл обычно возобновляется в течение 4 недель. Прямое действие на эндометрий приводит к специфичным для этого класса препаратов изменениям в эндометрии, связанным с антагонистическим действием на рецепторы прогестерона (Progesterone Receptor Modulator Associated Endometrial Changes (PAEC)). Как правило, гистологические изменения представлены неактивным и слабо пролиферирующим эпителием, сопровождающимся асимметрией роста стромы и эпителия, выраженным кистозным расширением желез со смешанными эстрогенными (митотическими) и прогестагенными (секреторными) влияниями на эпителий. Такие изменения отмечались примерно у 60% пациенток, получавших улипристал в течение 3 месяцев. Эти изменения обратимы и исчезают после прекращения лечения, их не следует принимать за гиперплазию эндометрия. Примерно у 5 % пациенток репродуктивного возраста с тяжелыми формами менструальных кровотечений толщина эндометрия больше 16 мм. У 10-15 % пациенток, получающих улипристал, эндометрий может утолщаться (>: 16 мм) в ходе лечения. Это утолщение исчезает после прекращения приема препарата и возобновления менструальных кровотечений. Если утолщение эндометрия сохраняется дольше 3 месяцев после окончания лечения и восстановления менструального цикла, следует провести дополнительное обследование для исключения других заболеваний. Лейомиома Улипристал оказывает прямое действие на лейомиомы, подавляя клеточную пролиферацию и индуцируя апоптоз, что приводит к уменьшению их размеров. Гипофиз При ежедневном приеме улипристала в дозе 5 мг происходит подавление овуляции у большинства пациенток, о чем свидетельствует поддержание концентрации прогестерона на уровне около 0,3 нг/мл. При ежедневном приеме улипристала в дозе 5 мг частично снижается концентрация фолликулостимулирующего гормона (ФСГ), однако концентрация эстрадиола в плазме крови у большинства пациенток поддерживается на уровне средней фолликулярной фазы и соответствует таковой в группе плацебо. Улипристал не влияет на концентрацию тироксинсвязывающего глобулина (ТСГ), адренокортикотропного гормона (АКТЕ) и пролактина в плазме крови на протяжении 3 месяцев лечения. Доклинические данные по безопасности В доклинических исследованиях фармакологической безопасности, токсичности многократных доз и генотоксичности потенциальных угроз для человека не выявлено. Основные находки в исследованиях общей токсичности связаны с влиянием на рецепторы прогестерона (а также на рецепторы глюкокортикостероидов при применении препарата в более высоких концентрациях), с антипрогестероновой активностью при экспозициях, близких к терапевтическим у человека. В 39-недельном исследовании на обезьянах с применением низких доз были выявлены изменения, сходные с РАЕС. В связи со своим механизмом действия улипристал вызывает гибель плода у крыс, кроликов (при многократных дозах выше 1 мг/кг), морских свинок и обезьян. Безопасность препарата в отношении эмбриона человека не установлена. В дозах, достаточно малых для сохранения беременности у животных, тератогенный потенциал не выявлен. В исследованиях репродукции у крыс с использованием доз, обеспечивающих такую же экспозицию, как у человека, не выявлено доказательств влияния на репродуктивную способность животных, получавших улипристал, а также на их потомство. В исследованиях, проведенных на мышах и крысах, канцерогенного действия улипристала не выявлено. Клиническая эффективности и безопасность Эффективность фиксированных доз улипристала 5 мг и 10 мг один раз в сутки оценивалась в двух исследованиях фазы 3, в которых участвовали пациентки с очень тяжелыми менструальными кровотечениями, вызванными миомой матки. В сравнении с плацебо было выявлено клинически значимое уменьшение объема менструальной кровопотери у пациенток, принимавших улипристал. Это позволяло быстро и более эффективно проводить коррекцию анемии, чем при назначении только препаратов железа. Уменьшение менструальной кровопотери у пациенток группы улипристала было сопоставимо с группой, получавшей агонист гонадотропин-рилизинг гормона (лейпрорелин). У большинства пациенток, получавших улипристал, кровотечение прекращалось в течение первой недели приема (развивалась аменорея). По данным магнитно-резонансной томографии в группе улипристала было значительно большее уменьшение размеров миомы матки, чем в группе плацебо. У пациенток, которым не была выполнена гистерэктомия или миомэктомия, при УЗ-контроле в конце лечения (неделя 13) оценивалось уменьшение размеров миомы матки. Как правило, оно сохранялось на протяжении 25 недель последующего наблюдения у пациенток группы улипристала, тогда как в группе, получавшей лейпрорелин, отмечалось некоторое увеличение размеров миомы матки. В рамках еще одного исследования фазы 3, в ходе которого пациентки получали по 2 курса терапии улипристалом 10 мг продолжительностью 3 месяца, частота аменореи была сопоставима в конце обоих курсов терапии. Уменьшение объема лейомиомы, зарегистрированное во время первого курса, сохранялось во время второго курса. С учетом результатов ранее проведенных исследований эффективность препарата в дозе 5 мг во время первого курса терапии будет такой же во время второго курса терапии, аналогично дозе 10 мг. Несмотря на ограниченное число пациенток, завершивших четыре 3-месячных курса лечения, полученных данных по безопасности достаточно для обоснования одного дополнительного 3-месячного курса терапии в предоперационном периоде. Фармакокинетика: Всасывание После однократного приема внутрь дозы 5 мг или 10 мг улипристал быстро всасывается, достигая примерно через 1 час после приема максимальной концентрации (Сmах) 23,5 ± 14,2 нг/мл и 50,0 ± 34,4 нг/мл соответственно. Площадь под кривой "концентрация-время" (AUC) составляет 61,3 ± 31,7 и 134,0 ± 83,8 нг-ч/мл соответственно. Улипристал быстро трансформируется в фармакологически активный метаболит, при этом через 1 час после приема Сmах составляет 9,0 ± 4,4 нг/мл и 20,6 ± 10,9 нг/мл, AUC 26,0 ± 12,0 и 63,6 ± 30,1 нг-ч/мл соответственно. Прием улипристала в дозе 30 мг вместе с завтраком с высоким содержанием жиров приводит к снижению средней Сmах примерно на 45 %, удлинению времени достижения максимальной концентрации (tmax) от медианы 0,75 ч до 3 ч и 25 % повышению AUC, по сравнению с приемом натощак. Такие же результаты получены для активного моно-N-деметилированного метаболита. Этот кинетический эффект пищи не расценивается как значимый для ежедневного приема таблеток улипристала. Распределение Улипристал в высокой степени (>: 98%) связывается с белками плазмы, включая альбумин, а-1-кислый гликопротеин, липопротеины высокой плотности и липопротеины низкой плотности. Улипристал и его активный N-деметилированный метаболит проникают в грудное молоко: среднее соотношение AUCt для молока/плазмы составляет 0,74 ± 0,32 для улипристала. Метаболизм Улипристал быстро превращается в vjyj-N-деметилированный и затем в ди-N- деметилированный метаболиты. Данные in vitro показывают, что этот процесс происходит в системе цитохрома Р450 с участием изофермента 3А4 (CYP3A4). Исходя из того, что метаболизм улипристала опосредован цитохромом Р450, ожидается влияние печtночной недостаточности на выведение улипристала, что приведет к увеличению его воздействия. Выведение Основной путь выведения - через кишечник, менее 10 % вещества выводится почками. Конечный период полувыведения улипристала после однократного приtма 5 мг или 10 мг составляет примерно 38 ч, средний клиренс около 100 л/ч. Данные in vitro показывают, что в клинически значимых концентрациях улипристал и его активный метаболит не ингибируют изоферменты CYP1A2, 2А6, 2С9, 2С19, 2D6, 2Е1 и ЗА4 и не индуцируют изофермент CYP1A2. Таким образом, применение улипристала не должно влиять на клиренс лекарственных препаратов, которые метаболизируются при участии данных изоферментов. Данные in vitro показывают, что улипристал и его активный метаболит не являются субстратами Р-гликопротеина (АВСВ1). Особые указания Улипристал назначается только после тщательного обследования. До начала лечения следует исключить беременность. Контрацепция В связи с возможностью нежелательных взаимодействий сопутствующее применение только гестагенсодержащих препаратов, гестагенвысвобождающих систем или комбинированных пероральных контрацептивов не рекомендуется. Хотя у большинства женщин, получавших терапевтические дозы улипристала, наблюдалась ановуляция, рекомендовано дополнительное использование негормонального метода контрацепции в ходе лечения. Почечная недостаточность Нет оснований предполагать, что почечная недостаточность может значительно влиять на выведение улипристала. Не рекомендуется применять улипристал без постоянного наблюдения у пациенток с тяжелой почечной недостаточностью, так как специальные исследования не проводились. Печеночная недостаточность Отсутствует опыт терапевтического применения улипристала у пациенток с печеночной недостаточностью. Ожидается, что печеночная недостаточность может влиять на выведение улипристала, что приведет к усилению действия препарата. Это несущественно для пациенток с печеночной недостаточностью легкой степени. Не рекомендуется назначать улипристал пациенткам с умеренной или тяжелой печеночной недостаточностью при невозможности постоянного наблюдения. Сопутствующая терапия Не рекомендуется сопутствующее применение улипристала и ингибиторов CYP3A4 средней мощности (например, эритромицина, грейпфрутового сока, верапамила) или мощных ингибиторов (например, кетоконазола, ритонавира, нефазодона, итраконазола, телитромицина, кларитромицина). Не рекомендуется совместное применение улипристала и мощных индукторов CYP3A4 (например, рифампицина, рифабутина, карбамазепина, окскарбазепина, фенитоина, фосфенитоина, фенобарбитала, примидона, зверобоя продырявленного, эфавиренза, невирапина, ритонавира - на фоне долгосрочного применения). Изменения эндометрия Улипристал оказывает специфическое фармакодинамическое действие на эндометрий. Может отмечаться увеличение толщины эндометрия. Если утолщение эндометрия сохраняется дольше 3 месяцев после окончания лечения и возобновления менструального цикла, следует провести дополнительное обследование для исключения других заболеваний. У пациенток, получающих улипристал, при гистологическом исследовании могут наблюдаться изменения строения эндометрия. Такие изменения обратимы после завершения лечения. Эти гистологические изменения обозначаются как изменения в эндометрии, связанные с антагонистическим действием на рецепторы прогестерона (РАЕС), и они не должны быть ошибочно оценены как гиперплазия эндометрия. Рекомендуется проводить не более 2 курсов терапии. Продолжительность каждого курса не должна превышать 3 месяцев, поскольку риск нежелательного воздействия на эндометрий на фоне более длительной терапии неизвестен. Кровотечение Пациентки должны быть проинформированы о том, что лечение улипристалом обычно приводит к значительному уменьшению менструальной кровопотери или аменорее в течение первых 10 дней лечения. При сохраняющихся чрезмерных кровотечениях пациентке следует обратиться к врачу. Как правило, менструальный цикл возобновляется в течение 4 недель после окончания курса лечения. Фертильность У большинства женщин, принимавших улипристал в терапевтических дозах, наблюдалась ановуляция. Однако фертильность при длительном применении улипристала не изучалась. Влияние на способность управлять транспортными средствами: Влияние на способность к управлению транспортными средствами и механизмами. Улипристал может оказывать минимальное влияние на способность к управлению транспортными средствами и механизмами, так как после приема улипристала может наблюдаться легкое головокружение. Условия хранения и отпуска из аптек Условия хранения:В защищенном от света месте, при температуре не выше 30 °C. Хранить в недоступном для детей месте. Отпуск из аптек: По рецепту Регистрационные данные Торговое название Эсмия® Международное непатентованное название:Улипристал. Форма выпуска:Таблетки. Состав:Состав на 1 таблетку: действующее вещество: улипристала ацетат 5,00 мг: вспомогательные вещества: целлюлоза микрокристаллическая 93,50 мг, маннитол 43,50 мг, тальк 4,00 мг, кроскармеллоза натрия 2,50 мг, магния стеарат 1,50 мг. АТХ: Регистрация: Лекарственное средство ЛП-002000 Фармгруппа: рецепторов прогестерона модулятор. Дата регистрации: 13.02.2013. Окончание регстрации: . Описание:Круглые двояковыпуклые таблетки белого или почти белого цвета с гравировкой "ES5" на одной стороне. Упаковка: Таблетки, 5 мг. По 14 таблеток в блистере из ПВХ/ПЭ/ПВДХ пленки оранжевого цвета и алюминиевой фольги. По 2 или 6 блистеров в картонную пачку вместе с инструкцией по применению. Срок годности:2 года. Не применять по истечении срока годности, указанного на упаковке. Владелец рег.удостоверения:ГЕДЕОН РИХТЕР, ОАО Производитель:GEDEON RICHTER, Ltd. Представительство:ГЕДЕОН РИХТЕР ОАО
10363 Руб.Розукард таб. п/о 40мг №90

Показания Первичная гиперхолестеринемия по классификации Фредриксона (тип IIа, включая семейную гетерозиготную гиперхолестеринемию) или смешанная гиперхолестеринемия (типа IIb) в качестве дополнения к диете, когда диета и другие немедикаментозные методы лечения (например, физические упражнения, снижение массы тела) оказываются недостаточными- Семейная гомозиготная гиперхолестеринемия в качестве дополнения к диете и другой липидснижающей терапии (например, ЛПНП-аферез), или в случаях, когда подобная терапия недостаточно эффективна- Гипертриглицеридемия (тип IV по классификации Фредриксона) в качестве дополнения к диете- Для замедления прогрессировать атеросклероза в качестве дополнения к диете у пациентов, которым показана терапия для снижения концентрации общего ХС и ХС -ЛПНП- Первичная профилактика основных сердечнососудистых осложнений (инсульта, инфаркта, артериальной реваскуляризации) у взрослых пациентов без клинических признаков ишемической болезни сердца (ИБС), по с повышенным риском се развития (возраст старше 50 лет для мужчин и старше 60 лет для женщин, повышенная концентрация С-реактивного белка (>: 2 мг/л) при наличии, как минимум одного из дополнительных факторов риска, таких как артериальная гипертензия, низкая концентрация ХС-ЛПВП, курение, семейный анамнез раннего начала ИБС). Противопоказания Противопоказания Противопоказания для таблеток 10 и 20 мг- Повышенная чувствительность к розувастатину или другим компонентам препарата- Заболевания печени в активной фазе или устойчивое повышение сывороточной активности "печеночных" трансаминаз (более чем в 3 раза по сравнению с верхней границей нормы) неясного генеза, печеночная недостаточность (степень тяжести от 7 до 9 баллов по шкале Чайлд-Пью)- Повышение концентрации креатининфосфокиназы (КФК) в крови более чем в 5 раз по. сравнению с верхней границей нормы (ВГН)- Наследственные заболевания, такие как непереносимость лактозы, дефицит лактазы или глкжозо-галактозная мальабсорбция (в связи с наличием в составе лактозы)- Выраженные нарушения функции почек (КК менее 30 мл/мин)- Миопатия: - Пациенты, предрасположенные к развитию миотоксических осложнений- Одновременный прием циклоспорина- Совместное применение с ингибиторами ВИЧ-протеаз- Женщины репродуктивного возраста, не пользующиеся адекватными методами контрацепции- Беременность и период лактации- Возраст до 18 лет (эффективность и безопасность не установлены). Противопоказания для таблеток 40 мг (дополнение к противопоказаниям для таблеток 10 и 20 мг)- Наличие следующих факторов риска развития миопатии/рабдомиолиза:1. Миотоксичность на фоне приема других ингибиторов ГМГ-КоА-редуктазы или фибратов в анамнезе: 2. Гипотиреоз:3. Почечная недостаточность средней степени тяжести (КК 30 - 60 мл/мин):4. Чрезмерное употребление алкоголя:5. Состояния, которые могут приводить к повышению плазменной концентрации розувастатина:6. Одновременный прием фибратов- Пациенты монголоидной расы- Семейный анамнез мышечных заболеваний. С осторожностью: Для дозировок 10 и 20мг: при заболеваниях печени в анамнезе: сепсисе: артериальной гипотензии: обширных хирургических вмешательствах: травмах: тяжелых метаболических, эндокринных или водно- электролитных нарушениях: неконтролируемых судорогах: при почечной недостаточности легкой и умеренной степени тяжести: гипотиреозе: данных о мышечной токсичности в анамнезе при применении других ингибиторов ГМГ-КоА- редуктазы или фибратов: наследственных мышечных заболеваниях в анамнезе: возрасте старше 65 лет: состояниях, при которых отмечено повышение концентрации розувастатина в плазме крови: применение у пациентов монголоидной расы: при одновременном назначении с фибратами: при чрезмерном употреблении алкоголя. Для дозировки 40 мг: при почечной недостаточности легкой степени тяжести (КК более 60 мл/мин): возрасте старше 65 лет: заболеваниях печени в анамнезе: сепсисе: артериальной гипотензии: обширных хирургических вмешательствах: травмах: тяжелых метаболических, эндокринных или водно- электролитных нарушениях: неконтролируемых судорогах. Беременность Применение препарата РОЗУКАРД® у женщин репродуктивного возраста возможно только в случае применения надежных методов контрацепции и если пациентка информирована о возможном риске лечения для плода. Поскольку холестерин и вещества, синтезируемые из холестерина важны для развития плода, потенциальный риск ингибирования ГМГ-КоА редуктазы превышает пользу от применения препарата во время беременности. РОЗУКАРД® противопоказан при беременности и в период лактации. В случае диагностирования беременности в процессе терапии препаратом прием РОЗУКАРДА® должен быть немедленно прекращен, а пациентка предупреждена о потенциальном риске для плода. При необходимости применения препарата в период лактации, учитывая возможность нежелательных явлений у грудных детей, следует решить вопрос о прекращении грудного вскармливания. Применение и дозы Внутрь, не разжевывая и не измельчая, проглатывать целиком, запивая водой, в любое время суток независимо от времени приема пищи. До начала терапии препаратом РОЗУКАРД® пациент должен начать соблюдать стандартную гиполипидемическую диету и продолжать соблюдать ее во время лечения. Дозу препарата следует подбирать индивидуально в зависимости от показаний и терапевтического ответа, принимая во внимание текущие общепринятые рекомендации по целевым концентрациям липидов. При необходимости приема препарата РОЗУКАРД® в дозе 5 мг следует разделить таблетку 10 мг на две части по риске. Рекомендуемая начальная доза препарата РОЗУКАРД® для пациентов, начинающих принимать препарат, или для пациентов, переведенных с приема других ингибиторов ГМГ-КоА-редуктазы, составляет 5 или 10 мг 1 раз в сутки. При выборе начальной дозы следует руководствоваться концентрацией холестерина в плазме крови и принимать во внимание риск развития у пациента сердечно-сосудистых осложнений, а также необходимо оценивать потенциальный риск развития побочных эффектов. В случае необходимости через 4 недели доза препарата может быть увеличена. В связи с возможным развитием побочных эффектов (см. раздел "Побочное действие") окончательное титрование до максимальной суточной дозы 40 мг следует проводить только у пациентов с тяжелой формой гиперхолестеринемии и высоким риском сердечно-сосудистых осложнений (особенно у пациентов с наследственной гиперхолестеринемией), у которых при приеме дозы в 20 мг не была достигнута целевая концентрация холестерина. Такие пациенты должны находиться под врачебным наблюдением. Рекомендуется особенно тщательное наблюдение за пациентами, получающими препарат в дозе 40 мг. Не рекомендуется назначение дозы 40 мг пациентам, ранее не обращавшимся к врачу. После 2-4-х недель терапии и/или при повышении дозы препарата РОЗУКАРД® необходимо проводить контроль показателей липидного обмена (при необходимости требуется коррекция дозы). Пожилые пациенты У пациентов старше 65 лет коррекции дозы не требуется. Пациенты с печеночной недостаточностью У пациентов с печеночной недостаточностью со значениями но шкале Чайлд-Пью ниже 7 баллов коррекции дозы препарата РОЗУКАРД® не требуется. Лекарственный препарат РОЗУКАРД® противопоказан пациентам с заболеваниями печени в активной фазе, устойчивым повышением сывороточной активности "печеночных" трансаминаз (более чем в 3 раза по сравнению с верхней границей нормы (ВГН)) неясного генеза и пациентам с печеночной недостаточностью (степень тяжести от 7 до 9 баллов по шкале Чайлд-Пью) (см. раздел "Противопоказания"). Пациенты с почечной недостаточностью У пациентов с почечной недостаточностью легкой степени тяжести коррекции дозы не требуется. Рекомендуется начальная доза препарата РОЗУКАРД® - 5 мг в сутки. У пациентов с почечной недостаточностью тяжелой степени тяжести (КК менее 30 мл/мин) применение препарата РОЗУКАРД® противопоказано. У пациентов с почечной недостаточностью умеренной степени тяжести (КК 30 - 60 мл/мин) применение препарата РОЗУКАРД® в дозе 40 мг в сутки противопоказано (см. раздел "Противопоказания). Особые популяции. Этнические группы При изучении фармакокинетических параметров розувастатина было отмечено увеличение его системной концентрации у представителей монголоидной расы (см. раздел "Фармакокинетика"). Следует учитывать данный факт при назначении препарата РОЗУКАРД® пациентам монголоидной расы. При назначении доз 10 и 20 мг рекомендуемая начальная доза препарата РОЗУКАРД® для данной группы пациентов составляет 5 мг в сутки. Применение препарата РОЗУКАРД® в дозе 40 мг в сутки у представителей монголоидной расы противопоказано (см. раздел "Противопоказания). Генетический полиморфизм У носителей генотипов SLCO1B1 (ОАТР1В1) с.521СС и ABCG2 (BCRP) с.421АА отмечалось увеличение экспозиции (AUC) к розувастатину по сравнению с носителями генотипов SLC01B1 C.521TT и ABCG2 С.421СС. Для пациентов-носителей генотипов с.521СС или с.421АА рекомендуемая максимальная доза препарата РОЗУКАРД® составляет 20 мг в сутки (см. разделы "Фармакокинетика", "Особые указания" и "Взаимодействие с другими лекарственными средствами"). Пациенты с предрасположенностью к миопатии Применение препарата РОЗУКАРД® в дозе 40 мг в сутки противопоказано у пациентов с предрасположенностью к миопатии (см. раздел "Противопоказания). При назначении доз 10 мг и 20 мг в сутки рекомендуемая начальная доза препарата РОЗУКАРД® для данной группы пациентов составляет 5 мг в сутки. Сопутствующая терапия Розувастатин связывается с различными транспортными белками (в частности, с ОАТР1В1 и BCRP). При совместном применении препарата РОЗУКАРД® с лекарственными препаратами (такими как циклоспорин, некоторые ингибиторы протеазы ВИЧ, включая комбинацию ритонавира с атазанавиром, лопинавиром и/или типранавиром), повышающими концентрацию розувастатина в плазме крови за счет взаимодействия с транспортными белками, может повышаться риск развития миопатии (включая рабдомиолиз) (см. разделы "Особые указания" и "Взаимодействие с другими лекарственными средствами"). В таких случаях следует оценить возможность назначения альтернативной терапии или временного прекращения приема препарата РОЗУКАРД®. Если же применение указанных выше препаратов необходимо, следует ознакомиться с инструкцией по применению препаратов перед их назначением одновременно с препаратом РОЗУКАРД®, оценить соотношение пользы и риска сопутствующей терапии и рассмотреть возможность снижения дозы препарата РОЗУКАРД® (см. раздел "Взаимодействие с другими лекарственными средствами"). Побочные эффекты и передозировка Побочные эффекты: Побочные эффекты. наблюдаемые при приеме розувастатина, обычно выражены незначительно и проходят самостоятельно. Как и при применении других ингибиторов ГМГ-КоА-редуктазы, частота возникновения побочных эффектов носит, в основном, дозозависимый характер. Ниже представлен профиль нежелательных реакций для розувастатина, составленный на основании данных из клинических исследований и обширного пострегистрационного опыта. Частота побочных реакций определялась соответственно следующей градации (классификация Всемирной Организации Здравоохранения)- очень часто - более 1/10- часто - от более 1/100 до менее 1/10- нечасто - от более 1/1000 до менее 1/100- редко - от более 1 /10000 до менее 1 /1000- очень редко - от менее 1/10000. включая отдельные сообщения- частота неизвестна. Не представляется возможным установить частоту по имеющимся данным. Нарушения со стороны крови и лимфатической системы: редко - тромбоцитопения. Нарушения со стороны нервной системы: часто - головная боль, головокружение: очень редко - периферическая пейропатия. потеря или снижение памяти: частота неизвестна - нарушения сна, включая бессоницу и "кошмарные " сновидения. Нарушения психики: частота неизвестна - депрессия. Нарушения со стороны пищеварительной системы: часто - тошнота, запор, боль в животе: нечасто - рвота: редко - панкреатит: частота неизвестна - диарея. Нарушения со стороны печени и желчевыводящих путей: редко - дозозависимое транзиторное повышение активности аспартатаминотрансферазы (ACT) и аланинаминотрансферазы (АЛТ): очень редко - гепатит, желтуха. Нарушения со стороны дыхательной системы: частота неизвестна - кашель, диспноэ. Нарушения со стороны иммунной системы: редко - реакции гиперчувствительности, включая ангионевротический отек. Нарушения со стороны скелетно-мышечной и соединительной ткани: часто - миалгия: редко - миопатия (включая миозит), рабдомиолиз с острой почечной недостаточностью и без нее (у пациентов, получавших лечение в дозах >: 20 мг в сутки): очень редко - артралгия: частота неизвестна - иммуноопосредованная некротизирующая миопатия. Нарушения со стороны кожи и подкожных тканей: нечасто - кожный зуд, крапивница, кожная сыпь: частота неизвестна - синдром Стивенса-Джонсона. Нарушения со стороны почек и мочевыводящих путей: часто - протеинурия. Изменения количества белка в моче (от отсутствия или следовых количеств до + или больше) наблюдаются у менее 1 % пациентов, получающих дозу препарата 10-20 мг в сутки, и у приблизительно 3 % пациентов, получающих 40 мг в сутки. Протеинурия уменьшается в процессе терапии и не связана с возникновением заболевания почек или инфекцией мочевыводящих путей: очень редко - гематурия. Нарушения со стороны половых органов и молочной железы: очень редко - гинекомастия. Влияние на показатели лабораторных и инструментальных исследований: нечасто - дозозависимое повышение активности сывороточной креатинфосфокиназы (КФК). В большинстве случаев оно было незначительным, бессимптомным и временным. При повышении активности более чем в 5 раз по сравнению с ВГН терапия препаратом РОЗУКАРД® должна быть временно приостановлена. Повышение концентрации гликозилированного гемоглобина в плазме крови. Прочие: часто - астенический синдром, частота неизвестна - периферические отеки. При применении препарата РОЗУКАРД® отмечались изменения следующих лабораторных показателей: повышение концентрации глюкозы, билирубина, активности щелочной фосфатазы, гамма-глутамилтрапсферазы (ГГТ). О развитии следующих нежелательных явлений сообщалось во время применения некоторых статинов: - эректильная дисфункция: - единичные случаи иитерстициалыюго заболевания легких (особенно при длительном применении)- сахарный диабет 2 типа: частота зависит от наличия или отсутствия факторов риска (концентрация глюкозы в крови натощак 5.6-6.9 ммоль/л, индекс массы тела (ИМ Г) >: 30 кг/м 2, гипертриглицеридемия, артериальная гипертензия в анамнезе). Передозировка: При одновременном приеме нескольких суточных доз фармакокинетические параметры розувастатина не меняются. Лечение: специфического лечения нет, проводится симптоматическая терапия для поддержания функций жизненно важных органов и систем. Необходим контроль показателей функции печени и активности КФК. Гемодиализ неэффективен. Взаимодействие с другими ЛС: Влияние применения других препаратов на розувастатин. Ингибиторы транспортных белков: розувастатин связывается с некоторыми транспортными белками, в частности, с ОАТР1В1 и BCRP. Сопутствующее применение препаратов, которые являются ингибиторами этих транспортных белков, может сопровождаться увеличением концентрации розувастатина в плазме крови и повышенным риском развития миопатии (см. таблицу 3 и разделы "Способ применения и дозы" и "Особые указания"). Циклоспорин: при одновременном применении розувастатина и циклоспорина AUC розувастатина была в среднем в 7 раз выше значения, которое отмечалось у здоровых добровольцев (см. таблицу 3). Не влияет на плазменную концентрацию циклоспорина. Препарат РОЗУКАРД® противопоказан пациентам, принимающим циклоспорин (см. раздел "Противопоказания"). Ингибиторы протеазы вируса иммунодефицита человека (ВИЧ): несмотря на то, что точный механизм взаимодействия неизвестен, совместный приём ингибиторов протеазы ВИЧ может приводить к значительному увеличению экспозиции розувастатина (см. табл. 3). Фармакокинетическое исследование по одновременному применению 20 мг розувастатина с комбинированным препаратом, содержащим два ингибитора протеазы ВИЧ (400 мг лопинавира/100 мг ритонавира) у здоровых добровольцев приводило к приблизительно двукратному и пятикратному увеличению AUC(0-24) и Сmах розувастатина, соответственно. Поэтому одновременный прием препарата РОЗУКАРД® и ингибиторов протеазы ВИЧ не рекомендуется (см. разделы "Способ применения и дозы", "Особые указания", таблицу 3). Гемфиброзил и другие гиполипидемические средства: совместное применение розувастатина и гемфиброзила приводит к увеличению в 2 раза Сmах и AUC розувастатина (см. раздел "Особые указания"). Основываясь на данных по специфическому взаимодействию, не ожидается фармакокинетически значимого взаимодействия с фенофибратом, фармакодинамическое взаимодействие возможно. Гемфиброзил, фенофибрат, другие фибраты и липидснижающис дозы никотиновой кислоты (более 1 г в сутки) увеличивали риск возникновения миопатии при одновременном применении с ингибиторами ГМГ-КоА- редуктазы, возможно в связи с тем, что они могут вызывать миоиатию при применении в монотерапии (см. раздел "Особые указания"). При одновременном приёме препарата с гемфиброзилом, фибратами, никотиновой кислотой в липидснижающих дозах пациентам рекомендуется начальная доза препарата РОЗУКАРД® 5 мг, прием в дозе 40 мг противопоказан при совместном назначении с фибратами (см. разделы "Противопоказания", "Способ применения и дозы", "Особые указания"). Фузидовая кислота: конкретных исследований но изучению лекарственного взаимодействия фузидовой кислоты и розувастатина не проводилось, но были отмечены отдельные сообщения о случаях рабдомиолиза. Эзетимиб: одновременное применение препарата РОЗУКАРД® в дозе 10 мг и эзетимиба в дозе 10 мг сопровождалось увеличением AUC розувастатина у пациентов с гиперхолестсринемией (см. таблицу 3). Нельзя исключить увеличение риска возникновения побочных эффектов из-за фармакодинамического взаимодействия между препаратом РОЗУКАРД® и эзетимибом. Антациды: одновременное применение розувастатина и суспензий антацидов, содержащих магния и алюминия гидроксид. приводит к снижению плазменной концентрации розувастатина примерно на 50 %. Данный эффект выражен слабее, если антациды применяются через 2 часа после приема розувастатина. Клиническое значение подобного взаимодействия не изучалось. Эритромицин: одновременное применение розувастатина и эритромицина приводит к уменьшению AUC розувастатина на 20 % и Сmax розувастатина на 30 %. Подобное взаимодействие может возникать в результате усиления моторики кишечника, вызываемого приёмом эритромицина. Изоферменты системы цитохрома 1450: результаты исследований in vivo и in vitro показали, что розувастатин не является ни ингибитором, ни индуктором изоферментов системы цитохрома Р450. Кроме того, розувастатин является слабым субстратом для этих изоферментов. Поэтому не ожидается взаимодействия розувастатина с другими лекарственными средствами на уровне метаболизма с участием изоферментов системы цитохрома Р450. Не отмечено клинически значимого взаимодействия розувастатина с флуконазолом (ингибитором изоферментов CYP2C9 и CYP3A4) и кетоконазолом (ингибитором изоферментов CYP2A6 и CYP3A4). Взаимодействие с лекарственными средствами, которое требует коррекции дозы розувастатина (см. таблицу 3) Дозу препарата РОЗУКАРД® следует корректировать при необходимости его совместного применения с лекарственными средствами, увеличивающими экспозицию розувастатина. Если ожидается увеличение экспозиции в 2 раза и более, начальная доза препарата РОЗУКАРД® должна составлять 5 мг один раз в сутки. Также следует корректировать максимальную суточную дозу препарата РОЗУКАРД® так, чтобы ожидаемая экспозиция розувастатина не превышала таковую для дозы 40 мг, принимаемой без одновременного назначения лекарственных средств. взаимодействующих с розувастатином. Например, максимальная суточная доза препарата РОЗУКАРД® при одновременном применении с гемфиброзилом составляет 20 мг (увеличение экспозиции в 1.9 раза), с ритонавиром/атазанавиром - 10 мг (увеличение экспозиции в 3.1 раза). Таблица 3. Влияние сопутствующей терапии на экспозицию розувастатина (AUC, данные приведены в порядке убывания) - результаты опубликованных клинических исследований. ТАБЛИЦЪ Влияние применения розувастатина на другие препараты. Антагонисты витамина К: начало терапии или увеличение дозы препарата РОЗУКЛРД® у пациентов, получающих одновременно антагонисты витамина К (например, варфарин), может приводить к увеличению Международного Нормализованного Отношения (MHO). Отмена или снижение дозы препарата РОЗУКАРД® может приводить к уменьшению MHO. В таких случаях рекомендуется контроль M1IO. Пероральные контрацептивы/гормонозаместительная терапия: одновременное применение розувастатина и пероральных контрацептивов увеличивает AUС этинилэстрадиола и AUC норгестрела на 26 % и 34 %, соответственно. Такое увеличение плазменной концентрации должно учитываться при подборе дозы пероральных контрацептивов. Фармакокинетические данные по одновременному применению препарата РОЗУКАРД® и гормонозаместительной терапии отсутствуют, следовательно, нельзя исключить аналогичного эффекта и при применении данного сочетания. Однако подобная комбинация широко применялась во время проведения клинических исследований и хорошо переносилась пациентами. Другие лекарственные средства: не ожидается клинически значимого взаимодействия розувастатина с дигоксином. Фармакологическое действие и фармакокинетика Гиполипидемическос средство из группы статинов. Селективный конкурентный ингибитор 3-гидрокси-3-метилглутарил коэнзим А (ГМГ-КоА)-редуктазы - фермента, превращающего ГМГ-КоА в мевалонат. предшественник холестерина (ХС). Увеличивает число рецепторов липопротеинов низкой плотности (ЛИНИ) на поверхности гепатоцитов, что приводит к усилению захвата и катаболизма ЛИНИ, ингибированию синтеза липопротеинов очень низкой плотности (ЛПОНП). уменьшая общую концентрацию ЛИНИ и ЛПОНП. Снижает концентрации ХС-ЛПНП, холсстерина-нелипопротеинов высокой плотности (ХС-неЛПВП), ХС-ЛПОНП, общего ХС, триглицеридов (ТГ), ТГ-ЛПОНП, аполипопрогеина В (АпоВ), снижает соотношения ХС-ЛПНП/ХС-ЛПВП, общий ХС/ХС-ЛПВП, ХС-неЛПВП/ХС-ЛПВП, АпоВ/аполипонротеина А1 (АпоА1), повышает концентрации ХС-ЛПВП и АпоА1. Гиполипидемическое действие прямо пропорционально величине назначенной дозы. Терапевтический эффект появляется в течение 1 недели после начала терапии, через 2 недели достигает 90 % от максимального, к 4 неделе достигает максимума и после этого остается постоянным. Таблица 1. Дозозависимый эффект у пациентов с первичной гиперхолестеринемией (тип На и lib по классификации Фредриксоиа) (среднее скорректированное процентное изменение по сравнению с исходным значением). ТАБЛИЦЪ Клиническая эффективность Эффективен у взрослых пациентов с гиперхолестеринемией с или без гипертриглицеридемии (вне зависимости от расы, пола или возраста), в том числе у пациентов с сахарным диабетом и семейной гиперхолестеринемией. У 80 % пациентов с гиперхолестеринемией IIа и IIb типа (по классификации Фредриксона) со средней исходной концентрацией ХС-ЛПНП около 4,8 ммоль/л на фоне приема препарата в дозе 10 мг концентрация ХС-ЛПНП достигает значений менее 3 ммоль/л. У пациентов с гетерозиготной семейной гиперхолестеринемией, получающих розувастатин в дозе 20-80 мг в сутки, отмечается положительная динамика показателей липидного профиля. После титрования до суточной дозы 40 мг (12 недель терапии), отмечалось снижение концентрации ХС-ЛПНП на 53 %. У 33 % пациентов достигалась концентрация ХС-ЛПНП менее 3 ммоль/л. У пациентов с гомозиготной семейной гиперхолестеринемией, принимающих препарат в дозе 20 мг и 40 мг, среднее снижение концентрации ХС-ЛПНП составляло 22 %. У пациентов с гипертриглицеридемией с начальной концентрацией ТГ от 273 до 817 мг/дл, получавших розувастатин в дозе от 5 мг до 40 мг один раз в сутки в течение 6-ти недель, значительно снижалась концентрация ТГ в плазме крови (см. таблицу 2). Аддитивный эффект отмечается в комбинации с фенофибратом (в отношении снижения концентрации ТГ и с никотиновой кислотой в липидснижающих дозах (более 1 г/сутки) (в отношении снижения концентрации ХС- ЛПВП). (см. раздел "Особые указания"). В исследовании METEOR терапия розувастатином значительно замедляла скорость прогрессирования максимальной толщины комплекса "интима-медиа" (ТКИМ) для 12 сегментов сонной артерии по сравнению с плацебо. По сравнению с исходными значениями в группе розувастатина было отмечено уменьшение максимального значения ТКИМ на 0,0014 мм/год по сравнению с увеличением этого показателя на 0.0131 мм/год в группе плацебо. До настоящего времени прямой зависимости между уменьшением ТКИМ и снижением риска сердечнососудистых событий продемонстрировано не было. Результаты проведенного исследования JUPITER показали, что розувастатин существенно снижал риск развития сердечно-сосудистых осложнений со снижением относительного риска на 44 %. Эффективность терапии была отмечена через 6 первых месяцев применения препарата. Отмечено статистически значимое снижение на 48 % комбинированного критерия, включавшего смерть от сердечно-сосудистых причин, инсульт и инфаркт миокарда, уменьшение па 54 % возникновения фатального или нефатального инфаркта миокарда и на 48 % - фатального или нефатального инсульта. Общая смертность снизилась на 20 % в группе розувастатина. Профиль безопасности у пациентов, принимавших розувастатин в дозе 20 мг, был. в целом, схож с профилем безопасности в группе плацебо. Фармакокинетика: Всасывание. Максимальная концентрация (Сmaх) розувастатина в плазме крови достигается приблизительно через 5 ч после приема препарата. Абсолютная биодоступиость - примерно 20 %. Распределение. Проникает через плацентарный барьер. Розувастатин поглощается приемущественно печенью, которая является основным местом синтеза ХС и метаболизма ХС-ЛПНП. Объем распределения - 134 л. Связывание с белками плазмы крови (преимущественно с альбумином) составляет приблизительно 90 %. Метаболизм. Биотрансформируется в печени в небольшой степени (около 10 %). являясь непрофильным субстратом для изоферментов системы цитохрома Р450. Основным изоферментом, участвующим в метаболизме розувастатина, является изофермент CYP2C9. Изоферменты CYP2C19, CYP3A4 и CYP2D6 вовлечены в метаболизм в меньшей степени. Основными метаболитами розувастатина являются N- дисметилрозувастатин и лактоновые метаболиты. N-дисметилрозувастатин примерно на 50 % менее активен, чем розувастатин, лактоновые метаболиты фармакологически неактивны. Более 90 % фармакологической активности по ингибироваиию циркулирующей ГМГ-КоА-редуктазы обеспечивается розувастатином, остальное - его метаболитами. Выведение. Около 90 % дозы розувастатина выводится в неизмененном виде через кишечник, оставшаяся часть - почками. Период полувыведения (Т1/2) - примерно 19 ч, не изменяется при увеличении дозы препарата. Среднее значение геометрического плазменного клиренса составляет приблизительно 50 л/ч (коэффициент вариации 21.7 %). Как и в случае других ингибиторов ГМГ-КоА-редуктазы в процесс печеночного захвата препарата вовлечен специфический мембранный переносчик - полипептид, транспортирующий органический анион ОАТР1В1. выполняющий важную роль в его печеночной элиминации. Линейность. Системная экспозиция розувастатина увеличивается пропорционально дозе. Фармакокинетические параметры не изменяются при ежедневном приеме. Особые популяции пациентов. Почечная недостаточностьУ пациентов с почечной недостаточностью легкой и умеренной степени тяжести плазменные концентрации розувастатина или N-дисметилрозувастатина существенно не меняются. У пациентов с тяжелой почечной недостаточностью (клиренс креатинина (КК) менее 30 мл/мин) концентрация розувастатина в плазме крови выше в 3 раза, а N-дисметилрозувастатина - в 9 раз, чем у здоровых добровольцев. Концентрация розувастатина в плазме крови у пациентов, находящихся на гемодиализе, примерно на 50 % выше, чем у здоровых добровольцев. Печеночная недостаточностьУ пациентов с баллом 7 и ниже по шкале Чайлд-Пью не выявлено увеличения T1/2 розувастатина: у пациентов с баллами 8 и 9 по шкале Чайлд-Пью отмечено удлинение T1/2 в 2 раза. Опыт применения препарата у пациентов с более выраженными нарушениями функции печени отсутствует. Пол и возраст не оказывают клинически значимого влияния на фармакокинетику розувастатина. Этнические группы. Фармакокинетические параметры розувастатина зависят от расовой принадлежности: площадь под кривой "концентрация-время" (AUC) у представителей монголоидной расы (японцы, китайцы, филиппинцы, вьетнамцы и корейцы) в 2 раза выше, чем у представителей европеоидной расы. У индусов среднее значение AUC и Сmах увеличено в 1.3 раза. Генетический полиморфизм Ингибиторы ГМГ-КоА-редуктазы, в том числе розувастатин связываются с транспортными белками ОАТР1В1 (полипептид транспорта органических анионов, участвующий в захвате статинов гепатоцитами) и BCRP (эффлюксиый транспортер). У носителей генотипов SLC01B1 (ОАТР1В1) с.521СС и ABCG2 (BCRP) c.421АА отмечалось увеличение экспозиции (AUC) к розувастатину в 1,6 и 2,4 раза, соответственно, по сравнению с носителями генотипов SLC01B1 c.521ТТ и ABCG2 с.421СС. Особые указания Почечные эффекты У пациентов, получавших высокие дозы розувастатина (и основном 40 мг), наблюдалась канальцевая протеинурия, которая в большинстве случаев была транзиторной. Такая протеинурия не свидетельствовала об остром заболевании почек или прогрессировании заболевания почек. У пациентов, принимающих препарат в дозе 40 мг, рекомендуется контролировать показатели функции почек во время лечения. Эффекты на опорно-двигательный аппарат При применении розувастатина во всех дозах и. в особенности при приеме доз препарата, превышающих 20 мг. сообщалось о следующих воздействиях на опорно- двигательный аппарат: миалгия. миопатия, в редких случаях рабдомиолиз. Определение активности креатинфосфокиназы (КФК) Определение активности КФК не следует проводить после интенсивных физических нагрузок или при наличии других возможных причин увеличения активности КФК, что может привести к неверной интерпретации полученных результатов. В случае если исходная активность КФК существенно повышена (в 5 раз выше ВГН), через 5-7 дней следует провести повторное измерение. Не следует начинать терапию, если повторный тест подтверждает исходную активность КФК (более чем в 5 раз выше ВГН). До начала терапии При применении препарата РОЗУКАРД®, также как и при применении других ингибиторов ГМГ-КоА-редуктазы, у пациентов с имеющимися факторами риска миопатии/рабдомиолиза следует проявлять осторожность (см. раздел "С осторожностью"). Следует оценить соотношение риска и пользы и в случае необходимости терапии проводить клиническое наблюдение за пациентом во время лечения. Во время терапии Следует проинформировать пациента о необходимости немедленного сообщения врачу о случаях неожиданного появления мышечных болей, мышечной слабости или спазмах, особенно в сочетании с недомоганием и лихорадкой. У таких пациентов следует определять активность КФК. Терапия должна быть прекращена, если активность КФК значительно повышена (более чем в 5 раз выше ВГН) или если симптомы со стороны мышц резко выражены и вызывают ежедневный дискомфорт (даже если активность КФК увеличена менее чем в 5 раз по сравнению с ВГН). Если симптомы исчезают, и активность КФК возвращается к норме, следует рассмотреть вопрос о повторном назначении препарата РОЗУКАРД® или других ингибиторов ГМГ-КоА-редуктазы в меньших дозах при тщательном наблюдении за пациентом. Рутинный контроль активности КФК при отсутствии симптомов нецелесообразен. Отмечены очень редкие случаи иммуноопосредованной некротизирующей миопатии с клиническими проявлениями в виде стойкой слабости проксимальных мышц и повышения активности КФК в сыворотке крови во время лечения или при прекращении приема статинов, в том числе розувастатина. Может потребоваться проведение дополнительных исследований мышечной и нервной системы, серологических исследований, а также терапия иммунодепрессивными средствами. Не отмечено признаков увеличения воздействия на скелетную мускулатуру при приеме розувастатина и сопутствующей терапии. Однако сообщалось об увеличении числа случаев миозита и миопатии у пациентов, принимавших другие ингибиторы ГМГ-КоА-редуктазы в сочетании с производными фибриновой кислоты, включая гемфиброзил, циклоспорин, никотиновую кислоту в липидснижающих дозах (более I г/сутки), азольные противогрибковые средства, ингибиторы протеазы ВИЧ и макролидные антибиотики. Гемфиброзил увеличивает риск возникновения миопатии при совместном применении с некоторыми ингибиторами ГМГ- КоА-редуктазы, Таким образом, не рекомендуется одновременное применение препарата РОЗУКАРД® и гемфиброзила. Должно быть тщательно взвешено соотношение риска и возможной пользы при совместном применении препарата РОЗУКАРД® и фибратов или липидснижающих доз никотиновой кислоты. Противопоказан прием препарата РОЗУКАРД® в дозе 40 мг совместно с фибратами (см. разделы "Взаимодействие с другими лекарственными средствами", "Противопоказания"). Через 2-4 недели после начала лечения и/или при повышении дозы препарата РОЗУКАРД® необходим контроль показателей липидного обмена (при необходимости требуется коррекция дозы). Печень Рекомендуется проводить определение показателей функции печени до начала терапии и через 3 месяца после начала терапии. Прием препарата РОЗУКАРД® следует прекратить или уменьшить дозу препарата, если активность "печеночных" трансаминаз в плазме крови в 3 раза превышает ВГН. У пациентов с гиперхолестеринемией вследствие гипотиреоза или нефротического синдрома терапия основных заболеваний должна проводиться до начала лечения препаратом РОЗУКАРД®. Особые популяции. Этнические группы В ходе фармакокинетических исследований среди китайских и японских пациентов отмечено увеличение системной концентрации розувастатина по сравнению с показателями, полученными среди пациентов - европеоидов (см. разделы "Способ применения и дозы" и "Фармакокииетика"). Ингибиторы протеазы ВИЧ Не рекомендуется совместное применение препарата РОЗУКАРД® с ингибиторами протеазы ВИЧ (см. раздел "Взаимодействие с другими лекарственными средствами"). Лактоза Препарат РОЗУКАРД® не следует применять у пациентов с лактазной недостаточностью, непереносимостью галактозы и глюкозо-галактозной мальабсорбнией. Интерстициальное заболевание легких При применении некоторых статинов, особенно в течение длительного времени, сообщалось о единичных случаях интерстициального заболевания легких. Проявлениями заболевания могут являться одышка, непродуктивный кашель и ухудшение общего самочувствия (слабость, снижение массы тела и лихорадка). При подозрении на интерстициальное заболевание легких необходимо прекратить терапию препаратом РОЗУКАРД®. Сахарный диабет 2 типа Препараты класса статинов способны вызывать повышение концентрации глюкозы в крови. У некоторых пациентов с высоким риском развития сахарного диабета такие изменения могут приводить к его манифестации, что является показанием для назначения гипогликемической терапии. Однако снижение риска сосудистых заболеваний на фоне приема статинов превышает риск развития сахарного диабета, поэтому данный фактор не должен служить основанием для отмены лечения статинами. За пациентами группы риска (концентрация глюкозы в крови натощак 5.6- 6,9 ммоль/л, индекс массы тела (ИМТ) >: 30 кг/м 2, гипертриглицеридемия, артериальная гипертепзия в анамнезе) следует установить врачебное наблюдение и регулярно проводить контроль биохимических параметров. Влияние на способность управлять транспортными средствами: Следует соблюдать осторожность при управлении транспортными средствами и занятиях, требующих повышенной концентрации внимания и быстроты психомоторных реакций (во время терапии может возникать головокружение). Условия хранения и отпуска из аптек Условия хранения:При температуре не выше 25 °С в оригинальной упаковке. Хранить в недоступном для детей месте! Отпуск из аптек: По рецепту Регистрационные данные Торговое название Розукард® Международное непатентованное название:Розувастатин. Форма выпуска:таблетки покрытые пленочной оболочкой. Состав:Каждая таблетка покрытая пленочной оболочкой 10 мг содержит: активное вещество: розувастатин - 10,000 мг (в виде розувастатина кальция - 10,400 мг): вспомогательные вещества: ядро: лактозы моногидрат - 60,000 мг, целлюлоза микрокристаллическая - 45,400 мг, кроскармеллоза натрия - 1,200 мг, кремния диоксид коллоидный - 0,600 мг, магния стеарат - 2,400 мг: пленочная оболочка: гипромеллоза 2910/5 - 2,500 мг, макрогол 6000 - 0,400 мг, титана диоксид - 0,325 мг, тальк - 0,475 мг, краситель железа оксид красный - 0,013 мг. Каждая таблетка покрытая пленочной оболочкой 20 мг содержит: активное вещество: розувастатин - 20,000 мг (в виде розувастатина кальция - 20,800 мг): вспомогательные вещества: ядро: лактозы моногидрат - 120,000 мг, целлюлоза микрокристаллическая - 90,800 мг, кроскармеллоза натрия - 2,400 мг, кремния диоксид коллоидный - 1,200 мг, магния стеарат - 4,800 мг: пленочная оболочка: гипромеллоза 2910/5 - 5,000 мг, макрогол 6000 - 0,800 мг, титана диоксид - 0,650 мг, тальк - 0,950 мг, краситель железа оксид красный - 0,065 мг. Каждая таблетка покрытая пленочной оболочкой 40 мг содержит: активное вещество: розувастатин - 40,000 мг (в виде розувастатина кальция - 41,600 мг): вспомогательные вещества: ядро: лактозы моногидрат - 240,000 мг, целлюлоза микрокристаллическая - 181,600 мг, кроскармеллоза натрия - 4,800 мг, кремния диоксид коллоидный - 2,400 мг, магния стеарат - 9,600 мг: пленочная оболочка: гипромеллоза 2910/5 - 10,000 мг, макрогол 6000 - 1,600 мг, титана диоксид - 1,300 мг, тальк - 1,900 мг, краситель железа оксид красный - 0,200 мг. АТХ: Регистрация: Лекарственное средство ЛП-001704 Фармгруппа: гиполипидемическое средство - ГМГ-КоА редуктазы ингибитор. Дата регистрации: 18.05.2012/29.01.2016. Окончание регстрации: 2017-05-18 2017-05-18 . Описание:Таблетки 10 мг: продолговатые, двояковыпуклые таблетки с риской, покрытые плёночной оболочкой светло-розового цвета. Таблетки 20 мг: продолговатые, двояковыпуклые таблетки, покрытые плёночной оболочкой розового цвета. Таблетки 40 мг: продолговатые, двояковыпуклые таблетки, покрытые плёночной оболочкой темно-розового цвета. Упаковка:Таблетки, покрытые пленочной оболочкой, 10 мг, 20 мг, 40 мг. По 10 таблеток в блистере из А1/А1. По 3, 6 или 9 блистеров (для дозировок 10 мг, 20 мг) и по 3 или 9 блистеров (для дозировки 40 мг) помещены в картонную пачку вместе с инструкцией по применению. Срок годности:2 года. Препарат нельзя применять после истечения срока годности, указанного на упаковке. Владелец рег.удостоверения:Зентива к.с. Производитель:ZENTIVA, k.s. Представительство*:ЗЕНТИВА ФАРМА, ООО
3879 Руб.Розукард таб. п/о 20мг №90
Показания Первичная гиперхолестеринемия по классификации Фредриксона (тип IIа, включая семейную гетерозиготную гиперхолестеринемию) или смешанная гиперхолестеринемия (типа IIb) в качестве дополнения к диете, когда диета и другие немедикаментозные методы лечения (например, физические упражнения, снижение массы тела) оказываются недостаточными- Семейная гомозиготная гиперхолестеринемия в качестве дополнения к диете и другой липидснижающей терапии (например, ЛПНП-аферез), или в случаях, когда подобная терапия недостаточно эффективна- Гипертриглицеридемия (тип IV по классификации Фредриксона) в качестве дополнения к диете- Для замедления прогрессировать атеросклероза в качестве дополнения к диете у пациентов, которым показана терапия для снижения концентрации общего ХС и ХС -ЛПНП- Первичная профилактика основных сердечнососудистых осложнений (инсульта, инфаркта, артериальной реваскуляризации) у взрослых пациентов без клинических признаков ишемической болезни сердца (ИБС), по с повышенным риском се развития (возраст старше 50 лет для мужчин и старше 60 лет для женщин, повышенная концентрация С-реактивного белка (>: 2 мг/л) при наличии, как минимум одного из дополнительных факторов риска, таких как артериальная гипертензия, низкая концентрация ХС-ЛПВП, курение, семейный анамнез раннего начала ИБС). Противопоказания Противопоказания Противопоказания для таблеток 10 и 20 мг- Повышенная чувствительность к розувастатину или другим компонентам препарата- Заболевания печени в активной фазе или устойчивое повышение сывороточной активности "печеночных" трансаминаз (более чем в 3 раза по сравнению с верхней границей нормы) неясного генеза, печеночная недостаточность (степень тяжести от 7 до 9 баллов по шкале Чайлд-Пью)- Повышение концентрации креатининфосфокиназы (КФК) в крови более чем в 5 раз по. сравнению с верхней границей нормы (ВГН)- Наследственные заболевания, такие как непереносимость лактозы, дефицит лактазы или глкжозо-галактозная мальабсорбция (в связи с наличием в составе лактозы)- Выраженные нарушения функции почек (КК менее 30 мл/мин)- Миопатия: - Пациенты, предрасположенные к развитию миотоксических осложнений- Одновременный прием циклоспорина- Совместное применение с ингибиторами ВИЧ-протеаз- Женщины репродуктивного возраста, не пользующиеся адекватными методами контрацепции- Беременность и период лактации- Возраст до 18 лет (эффективность и безопасность не установлены). Противопоказания для таблеток 40 мг (дополнение к противопоказаниям для таблеток 10 и 20 мг)- Наличие следующих факторов риска развития миопатии/рабдомиолиза:1. Миотоксичность на фоне приема других ингибиторов ГМГ-КоА-редуктазы или фибратов в анамнезе: 2. Гипотиреоз:3. Почечная недостаточность средней степени тяжести (КК 30 - 60 мл/мин):4. Чрезмерное употребление алкоголя:5. Состояния, которые могут приводить к повышению плазменной концентрации розувастатина:6. Одновременный прием фибратов- Пациенты монголоидной расы- Семейный анамнез мышечных заболеваний. С осторожностью: Для дозировок 10 и 20мг: при заболеваниях печени в анамнезе: сепсисе: артериальной гипотензии: обширных хирургических вмешательствах: травмах: тяжелых метаболических, эндокринных или водно- электролитных нарушениях: неконтролируемых судорогах: при почечной недостаточности легкой и умеренной степени тяжести: гипотиреозе: данных о мышечной токсичности в анамнезе при применении других ингибиторов ГМГ-КоА- редуктазы или фибратов: наследственных мышечных заболеваниях в анамнезе: возрасте старше 65 лет: состояниях, при которых отмечено повышение концентрации розувастатина в плазме крови: применение у пациентов монголоидной расы: при одновременном назначении с фибратами: при чрезмерном употреблении алкоголя. Для дозировки 40 мг: при почечной недостаточности легкой степени тяжести (КК более 60 мл/мин): возрасте старше 65 лет: заболеваниях печени в анамнезе: сепсисе: артериальной гипотензии: обширных хирургических вмешательствах: травмах: тяжелых метаболических, эндокринных или водно- электролитных нарушениях: неконтролируемых судорогах. Беременность Применение препарата РОЗУКАРД® у женщин репродуктивного возраста возможно только в случае применения надежных методов контрацепции и если пациентка информирована о возможном риске лечения для плода. Поскольку холестерин и вещества, синтезируемые из холестерина важны для развития плода, потенциальный риск ингибирования ГМГ-КоА редуктазы превышает пользу от применения препарата во время беременности. РОЗУКАРД® противопоказан при беременности и в период лактации. В случае диагностирования беременности в процессе терапии препаратом прием РОЗУКАРДА® должен быть немедленно прекращен, а пациентка предупреждена о потенциальном риске для плода. При необходимости применения препарата в период лактации, учитывая возможность нежелательных явлений у грудных детей, следует решить вопрос о прекращении грудного вскармливания. Применение и дозы Внутрь, не разжевывая и не измельчая, проглатывать целиком, запивая водой, в любое время суток независимо от времени приема пищи. До начала терапии препаратом РОЗУКАРД® пациент должен начать соблюдать стандартную гиполипидемическую диету и продолжать соблюдать ее во время лечения. Дозу препарата следует подбирать индивидуально в зависимости от показаний и терапевтического ответа, принимая во внимание текущие общепринятые рекомендации по целевым концентрациям липидов. При необходимости приема препарата РОЗУКАРД® в дозе 5 мг следует разделить таблетку 10 мг на две части по риске. Рекомендуемая начальная доза препарата РОЗУКАРД® для пациентов, начинающих принимать препарат, или для пациентов, переведенных с приема других ингибиторов ГМГ-КоА-редуктазы, составляет 5 или 10 мг 1 раз в сутки. При выборе начальной дозы следует руководствоваться концентрацией холестерина в плазме крови и принимать во внимание риск развития у пациента сердечно-сосудистых осложнений, а также необходимо оценивать потенциальный риск развития побочных эффектов. В случае необходимости через 4 недели доза препарата может быть увеличена. В связи с возможным развитием побочных эффектов (см. раздел "Побочное действие") окончательное титрование до максимальной суточной дозы 40 мг следует проводить только у пациентов с тяжелой формой гиперхолестеринемии и высоким риском сердечно-сосудистых осложнений (особенно у пациентов с наследственной гиперхолестеринемией), у которых при приеме дозы в 20 мг не была достигнута целевая концентрация холестерина. Такие пациенты должны находиться под врачебным наблюдением. Рекомендуется особенно тщательное наблюдение за пациентами, получающими препарат в дозе 40 мг. Не рекомендуется назначение дозы 40 мг пациентам, ранее не обращавшимся к врачу. После 2-4-х недель терапии и/или при повышении дозы препарата РОЗУКАРД® необходимо проводить контроль показателей липидного обмена (при необходимости требуется коррекция дозы). Пожилые пациенты У пациентов старше 65 лет коррекции дозы не требуется. Пациенты с печеночной недостаточностью У пациентов с печеночной недостаточностью со значениями но шкале Чайлд-Пью ниже 7 баллов коррекции дозы препарата РОЗУКАРД® не требуется. Лекарственный препарат РОЗУКАРД® противопоказан пациентам с заболеваниями печени в активной фазе, устойчивым повышением сывороточной активности "печеночных" трансаминаз (более чем в 3 раза по сравнению с верхней границей нормы (ВГН)) неясного генеза и пациентам с печеночной недостаточностью (степень тяжести от 7 до 9 баллов по шкале Чайлд-Пью) (см. раздел "Противопоказания"). Пациенты с почечной недостаточностью У пациентов с почечной недостаточностью легкой степени тяжести коррекции дозы не требуется. Рекомендуется начальная доза препарата РОЗУКАРД® - 5 мг в сутки. У пациентов с почечной недостаточностью тяжелой степени тяжести (КК менее 30 мл/мин) применение препарата РОЗУКАРД® противопоказано. У пациентов с почечной недостаточностью умеренной степени тяжести (КК 30 - 60 мл/мин) применение препарата РОЗУКАРД® в дозе 40 мг в сутки противопоказано (см. раздел "Противопоказания). Особые популяции. Этнические группы При изучении фармакокинетических параметров розувастатина было отмечено увеличение его системной концентрации у представителей монголоидной расы (см. раздел "Фармакокинетика"). Следует учитывать данный факт при назначении препарата РОЗУКАРД® пациентам монголоидной расы. При назначении доз 10 и 20 мг рекомендуемая начальная доза препарата РОЗУКАРД® для данной группы пациентов составляет 5 мг в сутки. Применение препарата РОЗУКАРД® в дозе 40 мг в сутки у представителей монголоидной расы противопоказано (см. раздел "Противопоказания). Генетический полиморфизм У носителей генотипов SLCO1B1 (ОАТР1В1) с.521СС и ABCG2 (BCRP) с.421АА отмечалось увеличение экспозиции (AUC) к розувастатину по сравнению с носителями генотипов SLC01B1 C.521TT и ABCG2 С.421СС. Для пациентов-носителей генотипов с.521СС или с.421АА рекомендуемая максимальная доза препарата РОЗУКАРД® составляет 20 мг в сутки (см. разделы "Фармакокинетика", "Особые указания" и "Взаимодействие с другими лекарственными средствами"). Пациенты с предрасположенностью к миопатии Применение препарата РОЗУКАРД® в дозе 40 мг в сутки противопоказано у пациентов с предрасположенностью к миопатии (см. раздел "Противопоказания). При назначении доз 10 мг и 20 мг в сутки рекомендуемая начальная доза препарата РОЗУКАРД® для данной группы пациентов составляет 5 мг в сутки. Сопутствующая терапия Розувастатин связывается с различными транспортными белками (в частности, с ОАТР1В1 и BCRP). При совместном применении препарата РОЗУКАРД® с лекарственными препаратами (такими как циклоспорин, некоторые ингибиторы протеазы ВИЧ, включая комбинацию ритонавира с атазанавиром, лопинавиром и/или типранавиром), повышающими концентрацию розувастатина в плазме крови за счет взаимодействия с транспортными белками, может повышаться риск развития миопатии (включая рабдомиолиз) (см. разделы "Особые указания" и "Взаимодействие с другими лекарственными средствами"). В таких случаях следует оценить возможность назначения альтернативной терапии или временного прекращения приема препарата РОЗУКАРД®. Если же применение указанных выше препаратов необходимо, следует ознакомиться с инструкцией по применению препаратов перед их назначением одновременно с препаратом РОЗУКАРД®, оценить соотношение пользы и риска сопутствующей терапии и рассмотреть возможность снижения дозы препарата РОЗУКАРД® (см. раздел "Взаимодействие с другими лекарственными средствами"). Побочные эффекты и передозировка Побочные эффекты: Побочные эффекты. наблюдаемые при приеме розувастатина, обычно выражены незначительно и проходят самостоятельно. Как и при применении других ингибиторов ГМГ-КоА-редуктазы, частота возникновения побочных эффектов носит, в основном, дозозависимый характер. Ниже представлен профиль нежелательных реакций для розувастатина, составленный на основании данных из клинических исследований и обширного пострегистрационного опыта. Частота побочных реакций определялась соответственно следующей градации (классификация Всемирной Организации Здравоохранения)- очень часто - более 1/10- часто - от более 1/100 до менее 1/10- нечасто - от более 1/1000 до менее 1/100- редко - от более 1 /10000 до менее 1 /1000- очень редко - от менее 1/10000. включая отдельные сообщения- частота неизвестна. Не представляется возможным установить частоту по имеющимся данным. Нарушения со стороны крови и лимфатической системы: редко - тромбоцитопения. Нарушения со стороны нервной системы: часто - головная боль, головокружение: очень редко - периферическая пейропатия. потеря или снижение памяти: частота неизвестна - нарушения сна, включая бессоницу и "кошмарные " сновидения. Нарушения психики: частота неизвестна - депрессия. Нарушения со стороны пищеварительной системы: часто - тошнота, запор, боль в животе: нечасто - рвота: редко - панкреатит: частота неизвестна - диарея. Нарушения со стороны печени и желчевыводящих путей: редко - дозозависимое транзиторное повышение активности аспартатаминотрансферазы (ACT) и аланинаминотрансферазы (АЛТ): очень редко - гепатит, желтуха. Нарушения со стороны дыхательной системы: частота неизвестна - кашель, диспноэ. Нарушения со стороны иммунной системы: редко - реакции гиперчувствительности, включая ангионевротический отек. Нарушения со стороны скелетно-мышечной и соединительной ткани: часто - миалгия: редко - миопатия (включая миозит), рабдомиолиз с острой почечной недостаточностью и без нее (у пациентов, получавших лечение в дозах >: 20 мг в сутки): очень редко - артралгия: частота неизвестна - иммуноопосредованная некротизирующая миопатия. Нарушения со стороны кожи и подкожных тканей: нечасто - кожный зуд, крапивница, кожная сыпь: частота неизвестна - синдром Стивенса-Джонсона. Нарушения со стороны почек и мочевыводящих путей: часто - протеинурия. Изменения количества белка в моче (от отсутствия или следовых количеств до + или больше) наблюдаются у менее 1 % пациентов, получающих дозу препарата 10-20 мг в сутки, и у приблизительно 3 % пациентов, получающих 40 мг в сутки. Протеинурия уменьшается в процессе терапии и не связана с возникновением заболевания почек или инфекцией мочевыводящих путей: очень редко - гематурия. Нарушения со стороны половых органов и молочной железы: очень редко - гинекомастия. Влияние на показатели лабораторных и инструментальных исследований: нечасто - дозозависимое повышение активности сывороточной креатинфосфокиназы (КФК). В большинстве случаев оно было незначительным, бессимптомным и временным. При повышении активности более чем в 5 раз по сравнению с ВГН терапия препаратом РОЗУКАРД® должна быть временно приостановлена. Повышение концентрации гликозилированного гемоглобина в плазме крови. Прочие: часто - астенический синдром, частота неизвестна - периферические отеки. При применении препарата РОЗУКАРД® отмечались изменения следующих лабораторных показателей: повышение концентрации глюкозы, билирубина, активности щелочной фосфатазы, гамма-глутамилтрапсферазы (ГГТ). О развитии следующих нежелательных явлений сообщалось во время применения некоторых статинов: - эректильная дисфункция: - единичные случаи иитерстициалыюго заболевания легких (особенно при длительном применении)- сахарный диабет 2 типа: частота зависит от наличия или отсутствия факторов риска (концентрация глюкозы в крови натощак 5.6-6.9 ммоль/л, индекс массы тела (ИМ Г) >: 30 кг/м 2, гипертриглицеридемия, артериальная гипертензия в анамнезе). Передозировка: При одновременном приеме нескольких суточных доз фармакокинетические параметры розувастатина не меняются. Лечение: специфического лечения нет, проводится симптоматическая терапия для поддержания функций жизненно важных органов и систем. Необходим контроль показателей функции печени и активности КФК. Гемодиализ неэффективен. Взаимодействие с другими ЛС: Влияние применения других препаратов на розувастатин. Ингибиторы транспортных белков: розувастатин связывается с некоторыми транспортными белками, в частности, с ОАТР1В1 и BCRP. Сопутствующее применение препаратов, которые являются ингибиторами этих транспортных белков, может сопровождаться увеличением концентрации розувастатина в плазме крови и повышенным риском развития миопатии (см. таблицу 3 и разделы "Способ применения и дозы" и "Особые указания"). Циклоспорин: при одновременном применении розувастатина и циклоспорина AUC розувастатина была в среднем в 7 раз выше значения, которое отмечалось у здоровых добровольцев (см. таблицу 3). Не влияет на плазменную концентрацию циклоспорина. Препарат РОЗУКАРД® противопоказан пациентам, принимающим циклоспорин (см. раздел "Противопоказания"). Ингибиторы протеазы вируса иммунодефицита человека (ВИЧ): несмотря на то, что точный механизм взаимодействия неизвестен, совместный приём ингибиторов протеазы ВИЧ может приводить к значительному увеличению экспозиции розувастатина (см. табл. 3). Фармакокинетическое исследование по одновременному применению 20 мг розувастатина с комбинированным препаратом, содержащим два ингибитора протеазы ВИЧ (400 мг лопинавира/100 мг ритонавира) у здоровых добровольцев приводило к приблизительно двукратному и пятикратному увеличению AUC(0-24) и Сmах розувастатина, соответственно. Поэтому одновременный прием препарата РОЗУКАРД® и ингибиторов протеазы ВИЧ не рекомендуется (см. разделы "Способ применения и дозы", "Особые указания", таблицу 3). Гемфиброзил и другие гиполипидемические средства: совместное применение розувастатина и гемфиброзила приводит к увеличению в 2 раза Сmах и AUC розувастатина (см. раздел "Особые указания"). Основываясь на данных по специфическому взаимодействию, не ожидается фармакокинетически значимого взаимодействия с фенофибратом, фармакодинамическое взаимодействие возможно. Гемфиброзил, фенофибрат, другие фибраты и липидснижающис дозы никотиновой кислоты (более 1 г в сутки) увеличивали риск возникновения миопатии при одновременном применении с ингибиторами ГМГ-КоА- редуктазы, возможно в связи с тем, что они могут вызывать миоиатию при применении в монотерапии (см. раздел "Особые указания"). При одновременном приёме препарата с гемфиброзилом, фибратами, никотиновой кислотой в липидснижающих дозах пациентам рекомендуется начальная доза препарата РОЗУКАРД® 5 мг, прием в дозе 40 мг противопоказан при совместном назначении с фибратами (см. разделы "Противопоказания", "Способ применения и дозы", "Особые указания"). Фузидовая кислота: конкретных исследований но изучению лекарственного взаимодействия фузидовой кислоты и розувастатина не проводилось, но были отмечены отдельные сообщения о случаях рабдомиолиза. Эзетимиб: одновременное применение препарата РОЗУКАРД® в дозе 10 мг и эзетимиба в дозе 10 мг сопровождалось увеличением AUC розувастатина у пациентов с гиперхолестсринемией (см. таблицу 3). Нельзя исключить увеличение риска возникновения побочных эффектов из-за фармакодинамического взаимодействия между препаратом РОЗУКАРД® и эзетимибом. Антациды: одновременное применение розувастатина и суспензий антацидов, содержащих магния и алюминия гидроксид. приводит к снижению плазменной концентрации розувастатина примерно на 50 %. Данный эффект выражен слабее, если антациды применяются через 2 часа после приема розувастатина. Клиническое значение подобного взаимодействия не изучалось. Эритромицин: одновременное применение розувастатина и эритромицина приводит к уменьшению AUC розувастатина на 20 % и Сmax розувастатина на 30 %. Подобное взаимодействие может возникать в результате усиления моторики кишечника, вызываемого приёмом эритромицина. Изоферменты системы цитохрома 1450: результаты исследований in vivo и in vitro показали, что розувастатин не является ни ингибитором, ни индуктором изоферментов системы цитохрома Р450. Кроме того, розувастатин является слабым субстратом для этих изоферментов. Поэтому не ожидается взаимодействия розувастатина с другими лекарственными средствами на уровне метаболизма с участием изоферментов системы цитохрома Р450. Не отмечено клинически значимого взаимодействия розувастатина с флуконазолом (ингибитором изоферментов CYP2C9 и CYP3A4) и кетоконазолом (ингибитором изоферментов CYP2A6 и CYP3A4). Взаимодействие с лекарственными средствами, которое требует коррекции дозы розувастатина (см. таблицу 3) Дозу препарата РОЗУКАРД® следует корректировать при необходимости его совместного применения с лекарственными средствами, увеличивающими экспозицию розувастатина. Если ожидается увеличение экспозиции в 2 раза и более, начальная доза препарата РОЗУКАРД® должна составлять 5 мг один раз в сутки. Также следует корректировать максимальную суточную дозу препарата РОЗУКАРД® так, чтобы ожидаемая экспозиция розувастатина не превышала таковую для дозы 40 мг, принимаемой без одновременного назначения лекарственных средств. взаимодействующих с розувастатином. Например, максимальная суточная доза препарата РОЗУКАРД® при одновременном применении с гемфиброзилом составляет 20 мг (увеличение экспозиции в 1.9 раза), с ритонавиром/атазанавиром - 10 мг (увеличение экспозиции в 3.1 раза). Таблица 3. Влияние сопутствующей терапии на экспозицию розувастатина (AUC, данные приведены в порядке убывания) - результаты опубликованных клинических исследований. ТАБЛИЦЪ Влияние применения розувастатина на другие препараты. Антагонисты витамина К: начало терапии или увеличение дозы препарата РОЗУКЛРД® у пациентов, получающих одновременно антагонисты витамина К (например, варфарин), может приводить к увеличению Международного Нормализованного Отношения (MHO). Отмена или снижение дозы препарата РОЗУКАРД® может приводить к уменьшению MHO. В таких случаях рекомендуется контроль M1IO. Пероральные контрацептивы/гормонозаместительная терапия: одновременное применение розувастатина и пероральных контрацептивов увеличивает AUС этинилэстрадиола и AUC норгестрела на 26 % и 34 %, соответственно. Такое увеличение плазменной концентрации должно учитываться при подборе дозы пероральных контрацептивов. Фармакокинетические данные по одновременному применению препарата РОЗУКАРД® и гормонозаместительной терапии отсутствуют, следовательно, нельзя исключить аналогичного эффекта и при применении данного сочетания. Однако подобная комбинация широко применялась во время проведения клинических исследований и хорошо переносилась пациентами. Другие лекарственные средства: не ожидается клинически значимого взаимодействия розувастатина с дигоксином. Фармакологическое действие и фармакокинетика Гиполипидемическос средство из группы статинов. Селективный конкурентный ингибитор 3-гидрокси-3-метилглутарил коэнзим А (ГМГ-КоА)-редуктазы - фермента, превращающего ГМГ-КоА в мевалонат. предшественник холестерина (ХС). Увеличивает число рецепторов липопротеинов низкой плотности (ЛИНИ) на поверхности гепатоцитов, что приводит к усилению захвата и катаболизма ЛИНИ, ингибированию синтеза липопротеинов очень низкой плотности (ЛПОНП). уменьшая общую концентрацию ЛИНИ и ЛПОНП. Снижает концентрации ХС-ЛПНП, холсстерина-нелипопротеинов высокой плотности (ХС-неЛПВП), ХС-ЛПОНП, общего ХС, триглицеридов (ТГ), ТГ-ЛПОНП, аполипопрогеина В (АпоВ), снижает соотношения ХС-ЛПНП/ХС-ЛПВП, общий ХС/ХС-ЛПВП, ХС-неЛПВП/ХС-ЛПВП, АпоВ/аполипонротеина А1 (АпоА1), повышает концентрации ХС-ЛПВП и АпоА1. Гиполипидемическое действие прямо пропорционально величине назначенной дозы. Терапевтический эффект появляется в течение 1 недели после начала терапии, через 2 недели достигает 90 % от максимального, к 4 неделе достигает максимума и после этого остается постоянным. Таблица 1. Дозозависимый эффект у пациентов с первичной гиперхолестеринемией (тип На и lib по классификации Фредриксоиа) (среднее скорректированное процентное изменение по сравнению с исходным значением). ТАБЛИЦЪ Клиническая эффективность Эффективен у взрослых пациентов с гиперхолестеринемией с или без гипертриглицеридемии (вне зависимости от расы, пола или возраста), в том числе у пациентов с сахарным диабетом и семейной гиперхолестеринемией. У 80 % пациентов с гиперхолестеринемией IIа и IIb типа (по классификации Фредриксона) со средней исходной концентрацией ХС-ЛПНП около 4,8 ммоль/л на фоне приема препарата в дозе 10 мг концентрация ХС-ЛПНП достигает значений менее 3 ммоль/л. У пациентов с гетерозиготной семейной гиперхолестеринемией, получающих розувастатин в дозе 20-80 мг в сутки, отмечается положительная динамика показателей липидного профиля. После титрования до суточной дозы 40 мг (12 недель терапии), отмечалось снижение концентрации ХС-ЛПНП на 53 %. У 33 % пациентов достигалась концентрация ХС-ЛПНП менее 3 ммоль/л. У пациентов с гомозиготной семейной гиперхолестеринемией, принимающих препарат в дозе 20 мг и 40 мг, среднее снижение концентрации ХС-ЛПНП составляло 22 %. У пациентов с гипертриглицеридемией с начальной концентрацией ТГ от 273 до 817 мг/дл, получавших розувастатин в дозе от 5 мг до 40 мг один раз в сутки в течение 6-ти недель, значительно снижалась концентрация ТГ в плазме крови (см. таблицу 2). Аддитивный эффект отмечается в комбинации с фенофибратом (в отношении снижения концентрации ТГ и с никотиновой кислотой в липидснижающих дозах (более 1 г/сутки) (в отношении снижения концентрации ХС- ЛПВП). (см. раздел "Особые указания"). В исследовании METEOR терапия розувастатином значительно замедляла скорость прогрессирования максимальной толщины комплекса "интима-медиа" (ТКИМ) для 12 сегментов сонной артерии по сравнению с плацебо. По сравнению с исходными значениями в группе розувастатина было отмечено уменьшение максимального значения ТКИМ на 0,0014 мм/год по сравнению с увеличением этого показателя на 0.0131 мм/год в группе плацебо. До настоящего времени прямой зависимости между уменьшением ТКИМ и снижением риска сердечнососудистых событий продемонстрировано не было. Результаты проведенного исследования JUPITER показали, что розувастатин существенно снижал риск развития сердечно-сосудистых осложнений со снижением относительного риска на 44 %. Эффективность терапии была отмечена через 6 первых месяцев применения препарата. Отмечено статистически значимое снижение на 48 % комбинированного критерия, включавшего смерть от сердечно-сосудистых причин, инсульт и инфаркт миокарда, уменьшение па 54 % возникновения фатального или нефатального инфаркта миокарда и на 48 % - фатального или нефатального инсульта. Общая смертность снизилась на 20 % в группе розувастатина. Профиль безопасности у пациентов, принимавших розувастатин в дозе 20 мг, был. в целом, схож с профилем безопасности в группе плацебо. Фармакокинетика: Всасывание. Максимальная концентрация (Сmaх) розувастатина в плазме крови достигается приблизительно через 5 ч после приема препарата. Абсолютная биодоступиость - примерно 20 %. Распределение. Проникает через плацентарный барьер. Розувастатин поглощается приемущественно печенью, которая является основным местом синтеза ХС и метаболизма ХС-ЛПНП. Объем распределения - 134 л. Связывание с белками плазмы крови (преимущественно с альбумином) составляет приблизительно 90 %. Метаболизм. Биотрансформируется в печени в небольшой степени (около 10 %). являясь непрофильным субстратом для изоферментов системы цитохрома Р450. Основным изоферментом, участвующим в метаболизме розувастатина, является изофермент CYP2C9. Изоферменты CYP2C19, CYP3A4 и CYP2D6 вовлечены в метаболизм в меньшей степени. Основными метаболитами розувастатина являются N- дисметилрозувастатин и лактоновые метаболиты. N-дисметилрозувастатин примерно на 50 % менее активен, чем розувастатин, лактоновые метаболиты фармакологически неактивны. Более 90 % фармакологической активности по ингибироваиию циркулирующей ГМГ-КоА-редуктазы обеспечивается розувастатином, остальное - его метаболитами. Выведение. Около 90 % дозы розувастатина выводится в неизмененном виде через кишечник, оставшаяся часть - почками. Период полувыведения (Т1/2) - примерно 19 ч, не изменяется при увеличении дозы препарата. Среднее значение геометрического плазменного клиренса составляет приблизительно 50 л/ч (коэффициент вариации 21.7 %). Как и в случае других ингибиторов ГМГ-КоА-редуктазы в процесс печеночного захвата препарата вовлечен специфический мембранный переносчик - полипептид, транспортирующий органический анион ОАТР1В1. выполняющий важную роль в его печеночной элиминации. Линейность. Системная экспозиция розувастатина увеличивается пропорционально дозе. Фармакокинетические параметры не изменяются при ежедневном приеме. Особые популяции пациентов. Почечная недостаточностьУ пациентов с почечной недостаточностью легкой и умеренной степени тяжести плазменные концентрации розувастатина или N-дисметилрозувастатина существенно не меняются. У пациентов с тяжелой почечной недостаточностью (клиренс креатинина (КК) менее 30 мл/мин) концентрация розувастатина в плазме крови выше в 3 раза, а N-дисметилрозувастатина - в 9 раз, чем у здоровых добровольцев. Концентрация розувастатина в плазме крови у пациентов, находящихся на гемодиализе, примерно на 50 % выше, чем у здоровых добровольцев. Печеночная недостаточностьУ пациентов с баллом 7 и ниже по шкале Чайлд-Пью не выявлено увеличения T1/2 розувастатина: у пациентов с баллами 8 и 9 по шкале Чайлд-Пью отмечено удлинение T1/2 в 2 раза. Опыт применения препарата у пациентов с более выраженными нарушениями функции печени отсутствует. Пол и возраст не оказывают клинически значимого влияния на фармакокинетику розувастатина. Этнические группы. Фармакокинетические параметры розувастатина зависят от расовой принадлежности: площадь под кривой "концентрация-время" (AUC) у представителей монголоидной расы (японцы, китайцы, филиппинцы, вьетнамцы и корейцы) в 2 раза выше, чем у представителей европеоидной расы. У индусов среднее значение AUC и Сmах увеличено в 1.3 раза. Генетический полиморфизм Ингибиторы ГМГ-КоА-редуктазы, в том числе розувастатин связываются с транспортными белками ОАТР1В1 (полипептид транспорта органических анионов, участвующий в захвате статинов гепатоцитами) и BCRP (эффлюксиый транспортер). У носителей генотипов SLC01B1 (ОАТР1В1) с.521СС и ABCG2 (BCRP) c.421АА отмечалось увеличение экспозиции (AUC) к розувастатину в 1,6 и 2,4 раза, соответственно, по сравнению с носителями генотипов SLC01B1 c.521ТТ и ABCG2 с.421СС. Особые указания Почечные эффекты У пациентов, получавших высокие дозы розувастатина (и основном 40 мг), наблюдалась канальцевая протеинурия, которая в большинстве случаев была транзиторной. Такая протеинурия не свидетельствовала об остром заболевании почек или прогрессировании заболевания почек. У пациентов, принимающих препарат в дозе 40 мг, рекомендуется контролировать показатели функции почек во время лечения. Эффекты на опорно-двигательный аппарат При применении розувастатина во всех дозах и. в особенности при приеме доз препарата, превышающих 20 мг. сообщалось о следующих воздействиях на опорно- двигательный аппарат: миалгия. миопатия, в редких случаях рабдомиолиз. Определение активности креатинфосфокиназы (КФК) Определение активности КФК не следует проводить после интенсивных физических нагрузок или при наличии других возможных причин увеличения активности КФК, что может привести к неверной интерпретации полученных результатов. В случае если исходная активность КФК существенно повышена (в 5 раз выше ВГН), через 5-7 дней следует провести повторное измерение. Не следует начинать терапию, если повторный тест подтверждает исходную активность КФК (более чем в 5 раз выше ВГН). До начала терапии При применении препарата РОЗУКАРД®, также как и при применении других ингибиторов ГМГ-КоА-редуктазы, у пациентов с имеющимися факторами риска миопатии/рабдомиолиза следует проявлять осторожность (см. раздел "С осторожностью"). Следует оценить соотношение риска и пользы и в случае необходимости терапии проводить клиническое наблюдение за пациентом во время лечения. Во время терапии Следует проинформировать пациента о необходимости немедленного сообщения врачу о случаях неожиданного появления мышечных болей, мышечной слабости или спазмах, особенно в сочетании с недомоганием и лихорадкой. У таких пациентов следует определять активность КФК. Терапия должна быть прекращена, если активность КФК значительно повышена (более чем в 5 раз выше ВГН) или если симптомы со стороны мышц резко выражены и вызывают ежедневный дискомфорт (даже если активность КФК увеличена менее чем в 5 раз по сравнению с ВГН). Если симптомы исчезают, и активность КФК возвращается к норме, следует рассмотреть вопрос о повторном назначении препарата РОЗУКАРД® или других ингибиторов ГМГ-КоА-редуктазы в меньших дозах при тщательном наблюдении за пациентом. Рутинный контроль активности КФК при отсутствии симптомов нецелесообразен. Отмечены очень редкие случаи иммуноопосредованной некротизирующей миопатии с клиническими проявлениями в виде стойкой слабости проксимальных мышц и повышения активности КФК в сыворотке крови во время лечения или при прекращении приема статинов, в том числе розувастатина. Может потребоваться проведение дополнительных исследований мышечной и нервной системы, серологических исследований, а также терапия иммунодепрессивными средствами. Не отмечено признаков увеличения воздействия на скелетную мускулатуру при приеме розувастатина и сопутствующей терапии. Однако сообщалось об увеличении числа случаев миозита и миопатии у пациентов, принимавших другие ингибиторы ГМГ-КоА-редуктазы в сочетании с производными фибриновой кислоты, включая гемфиброзил, циклоспорин, никотиновую кислоту в липидснижающих дозах (более I г/сутки), азольные противогрибковые средства, ингибиторы протеазы ВИЧ и макролидные антибиотики. Гемфиброзил увеличивает риск возникновения миопатии при совместном применении с некоторыми ингибиторами ГМГ- КоА-редуктазы, Таким образом, не рекомендуется одновременное применение препарата РОЗУКАРД® и гемфиброзила. Должно быть тщательно взвешено соотношение риска и возможной пользы при совместном применении препарата РОЗУКАРД® и фибратов или липидснижающих доз никотиновой кислоты. Противопоказан прием препарата РОЗУКАРД® в дозе 40 мг совместно с фибратами (см. разделы "Взаимодействие с другими лекарственными средствами", "Противопоказания"). Через 2-4 недели после начала лечения и/или при повышении дозы препарата РОЗУКАРД® необходим контроль показателей липидного обмена (при необходимости требуется коррекция дозы). Печень Рекомендуется проводить определение показателей функции печени до начала терапии и через 3 месяца после начала терапии. Прием препарата РОЗУКАРД® следует прекратить или уменьшить дозу препарата, если активность "печеночных" трансаминаз в плазме крови в 3 раза превышает ВГН. У пациентов с гиперхолестеринемией вследствие гипотиреоза или нефротического синдрома терапия основных заболеваний должна проводиться до начала лечения препаратом РОЗУКАРД®. Особые популяции. Этнические группы В ходе фармакокинетических исследований среди китайских и японских пациентов отмечено увеличение системной концентрации розувастатина по сравнению с показателями, полученными среди пациентов - европеоидов (см. разделы "Способ применения и дозы" и "Фармакокииетика"). Ингибиторы протеазы ВИЧ Не рекомендуется совместное применение препарата РОЗУКАРД® с ингибиторами протеазы ВИЧ (см. раздел "Взаимодействие с другими лекарственными средствами"). Лактоза Препарат РОЗУКАРД® не следует применять у пациентов с лактазной недостаточностью, непереносимостью галактозы и глюкозо-галактозной мальабсорбнией. Интерстициальное заболевание легких При применении некоторых статинов, особенно в течение длительного времени, сообщалось о единичных случаях интерстициального заболевания легких. Проявлениями заболевания могут являться одышка, непродуктивный кашель и ухудшение общего самочувствия (слабость, снижение массы тела и лихорадка). При подозрении на интерстициальное заболевание легких необходимо прекратить терапию препаратом РОЗУКАРД®. Сахарный диабет 2 типа Препараты класса статинов способны вызывать повышение концентрации глюкозы в крови. У некоторых пациентов с высоким риском развития сахарного диабета такие изменения могут приводить к его манифестации, что является показанием для назначения гипогликемической терапии. Однако снижение риска сосудистых заболеваний на фоне приема статинов превышает риск развития сахарного диабета, поэтому данный фактор не должен служить основанием для отмены лечения статинами. За пациентами группы риска (концентрация глюкозы в крови натощак 5.6- 6,9 ммоль/л, индекс массы тела (ИМТ) >: 30 кг/м 2, гипертриглицеридемия, артериальная гипертепзия в анамнезе) следует установить врачебное наблюдение и регулярно проводить контроль биохимических параметров. Влияние на способность управлять транспортными средствами: Следует соблюдать осторожность при управлении транспортными средствами и занятиях, требующих повышенной концентрации внимания и быстроты психомоторных реакций (во время терапии может возникать головокружение). Условия хранения и отпуска из аптек Условия хранения:При температуре не выше 25 °С в оригинальной упаковке. Хранить в недоступном для детей месте! Отпуск из аптек: По рецепту Регистрационные данные Торговое название Розукард® Международное непатентованное название:Розувастатин. Форма выпуска:таблетки покрытые пленочной оболочкой. Состав:Каждая таблетка покрытая пленочной оболочкой 10 мг содержит: активное вещество: розувастатин - 10,000 мг (в виде розувастатина кальция - 10,400 мг): вспомогательные вещества: ядро: лактозы моногидрат - 60,000 мг, целлюлоза микрокристаллическая - 45,400 мг, кроскармеллоза натрия - 1,200 мг, кремния диоксид коллоидный - 0,600 мг, магния стеарат - 2,400 мг: пленочная оболочка: гипромеллоза 2910/5 - 2,500 мг, макрогол 6000 - 0,400 мг, титана диоксид - 0,325 мг, тальк - 0,475 мг, краситель железа оксид красный - 0,013 мг. Каждая таблетка покрытая пленочной оболочкой 20 мг содержит: активное вещество: розувастатин - 20,000 мг (в виде розувастатина кальция - 20,800 мг): вспомогательные вещества: ядро: лактозы моногидрат - 120,000 мг, целлюлоза микрокристаллическая - 90,800 мг, кроскармеллоза натрия - 2,400 мг, кремния диоксид коллоидный - 1,200 мг, магния стеарат - 4,800 мг: пленочная оболочка: гипромеллоза 2910/5 - 5,000 мг, макрогол 6000 - 0,800 мг, титана диоксид - 0,650 мг, тальк - 0,950 мг, краситель железа оксид красный - 0,065 мг. Каждая таблетка покрытая пленочной оболочкой 40 мг содержит: активное вещество: розувастатин - 40,000 мг (в виде розувастатина кальция - 41,600 мг): вспомогательные вещества: ядро: лактозы моногидрат - 240,000 мг, целлюлоза микрокристаллическая - 181,600 мг, кроскармеллоза натрия - 4,800 мг, кремния диоксид коллоидный - 2,400 мг, магния стеарат - 9,600 мг: пленочная оболочка: гипромеллоза 2910/5 - 10,000 мг, макрогол 6000 - 1,600 мг, титана диоксид - 1,300 мг, тальк - 1,900 мг, краситель железа оксид красный - 0,200 мг. АТХ: Регистрация: Лекарственное средство ЛП-001704 Фармгруппа: гиполипидемическое средство - ГМГ-КоА редуктазы ингибитор. Дата регистрации: 18.05.2012/29.01.2016. Окончание регстрации: 2017-05-18 2017-05-18 . Описание:Таблетки 10 мг: продолговатые, двояковыпуклые таблетки с риской, покрытые плёночной оболочкой светло-розового цвета. Таблетки 20 мг: продолговатые, двояковыпуклые таблетки, покрытые плёночной оболочкой розового цвета. Таблетки 40 мг: продолговатые, двояковыпуклые таблетки, покрытые плёночной оболочкой темно-розового цвета. Упаковка:Таблетки, покрытые пленочной оболочкой, 10 мг, 20 мг, 40 мг. По 10 таблеток в блистере из А1/А1. По 3, 6 или 9 блистеров (для дозировок 10 мг, 20 мг) и по 3 или 9 блистеров (для дозировки 40 мг) помещены в картонную пачку вместе с инструкцией по применению. Срок годности:2 года. Препарат нельзя применять после истечения срока годности, указанного на упаковке. Владелец рег.удостоверения:Зентива к.с. Производитель:ZENTIVA, k.s. Представительство*:ЗЕНТИВА ФАРМА, ООО
2039 Руб.Розукард таб. п/о 20мг №30
Показания Первичная гиперхолестеринемия по классификации Фредриксона (тип IIа, включая семейную гетерозиготную гиперхолестеринемию) или смешанная гиперхолестеринемия (типа IIb) в качестве дополнения к диете, когда диета и другие немедикаментозные методы лечения (например, физические упражнения, снижение массы тела) оказываются недостаточными- Семейная гомозиготная гиперхолестеринемия в качестве дополнения к диете и другой липидснижающей терапии (например, ЛПНП-аферез), или в случаях, когда подобная терапия недостаточно эффективна- Гипертриглицеридемия (тип IV по классификации Фредриксона) в качестве дополнения к диете- Для замедления прогрессировать атеросклероза в качестве дополнения к диете у пациентов, которым показана терапия для снижения концентрации общего ХС и ХС -ЛПНП- Первичная профилактика основных сердечнососудистых осложнений (инсульта, инфаркта, артериальной реваскуляризации) у взрослых пациентов без клинических признаков ишемической болезни сердца (ИБС), по с повышенным риском се развития (возраст старше 50 лет для мужчин и старше 60 лет для женщин, повышенная концентрация С-реактивного белка (>: 2 мг/л) при наличии, как минимум одного из дополнительных факторов риска, таких как артериальная гипертензия, низкая концентрация ХС-ЛПВП, курение, семейный анамнез раннего начала ИБС). Противопоказания Противопоказания Противопоказания для таблеток 10 и 20 мг- Повышенная чувствительность к розувастатину или другим компонентам препарата- Заболевания печени в активной фазе или устойчивое повышение сывороточной активности "печеночных" трансаминаз (более чем в 3 раза по сравнению с верхней границей нормы) неясного генеза, печеночная недостаточность (степень тяжести от 7 до 9 баллов по шкале Чайлд-Пью)- Повышение концентрации креатининфосфокиназы (КФК) в крови более чем в 5 раз по. сравнению с верхней границей нормы (ВГН)- Наследственные заболевания, такие как непереносимость лактозы, дефицит лактазы или глкжозо-галактозная мальабсорбция (в связи с наличием в составе лактозы)- Выраженные нарушения функции почек (КК менее 30 мл/мин)- Миопатия: - Пациенты, предрасположенные к развитию миотоксических осложнений- Одновременный прием циклоспорина- Совместное применение с ингибиторами ВИЧ-протеаз- Женщины репродуктивного возраста, не пользующиеся адекватными методами контрацепции- Беременность и период лактации- Возраст до 18 лет (эффективность и безопасность не установлены). Противопоказания для таблеток 40 мг (дополнение к противопоказаниям для таблеток 10 и 20 мг)- Наличие следующих факторов риска развития миопатии/рабдомиолиза:1. Миотоксичность на фоне приема других ингибиторов ГМГ-КоА-редуктазы или фибратов в анамнезе: 2. Гипотиреоз:3. Почечная недостаточность средней степени тяжести (КК 30 - 60 мл/мин):4. Чрезмерное употребление алкоголя:5. Состояния, которые могут приводить к повышению плазменной концентрации розувастатина:6. Одновременный прием фибратов- Пациенты монголоидной расы- Семейный анамнез мышечных заболеваний. С осторожностью: Для дозировок 10 и 20мг: при заболеваниях печени в анамнезе: сепсисе: артериальной гипотензии: обширных хирургических вмешательствах: травмах: тяжелых метаболических, эндокринных или водно- электролитных нарушениях: неконтролируемых судорогах: при почечной недостаточности легкой и умеренной степени тяжести: гипотиреозе: данных о мышечной токсичности в анамнезе при применении других ингибиторов ГМГ-КоА- редуктазы или фибратов: наследственных мышечных заболеваниях в анамнезе: возрасте старше 65 лет: состояниях, при которых отмечено повышение концентрации розувастатина в плазме крови: применение у пациентов монголоидной расы: при одновременном назначении с фибратами: при чрезмерном употреблении алкоголя. Для дозировки 40 мг: при почечной недостаточности легкой степени тяжести (КК более 60 мл/мин): возрасте старше 65 лет: заболеваниях печени в анамнезе: сепсисе: артериальной гипотензии: обширных хирургических вмешательствах: травмах: тяжелых метаболических, эндокринных или водно- электролитных нарушениях: неконтролируемых судорогах. Беременность Применение препарата РОЗУКАРД® у женщин репродуктивного возраста возможно только в случае применения надежных методов контрацепции и если пациентка информирована о возможном риске лечения для плода. Поскольку холестерин и вещества, синтезируемые из холестерина важны для развития плода, потенциальный риск ингибирования ГМГ-КоА редуктазы превышает пользу от применения препарата во время беременности. РОЗУКАРД® противопоказан при беременности и в период лактации. В случае диагностирования беременности в процессе терапии препаратом прием РОЗУКАРДА® должен быть немедленно прекращен, а пациентка предупреждена о потенциальном риске для плода. При необходимости применения препарата в период лактации, учитывая возможность нежелательных явлений у грудных детей, следует решить вопрос о прекращении грудного вскармливания. Применение и дозы Внутрь, не разжевывая и не измельчая, проглатывать целиком, запивая водой, в любое время суток независимо от времени приема пищи. До начала терапии препаратом РОЗУКАРД® пациент должен начать соблюдать стандартную гиполипидемическую диету и продолжать соблюдать ее во время лечения. Дозу препарата следует подбирать индивидуально в зависимости от показаний и терапевтического ответа, принимая во внимание текущие общепринятые рекомендации по целевым концентрациям липидов. При необходимости приема препарата РОЗУКАРД® в дозе 5 мг следует разделить таблетку 10 мг на две части по риске. Рекомендуемая начальная доза препарата РОЗУКАРД® для пациентов, начинающих принимать препарат, или для пациентов, переведенных с приема других ингибиторов ГМГ-КоА-редуктазы, составляет 5 или 10 мг 1 раз в сутки. При выборе начальной дозы следует руководствоваться концентрацией холестерина в плазме крови и принимать во внимание риск развития у пациента сердечно-сосудистых осложнений, а также необходимо оценивать потенциальный риск развития побочных эффектов. В случае необходимости через 4 недели доза препарата может быть увеличена. В связи с возможным развитием побочных эффектов (см. раздел "Побочное действие") окончательное титрование до максимальной суточной дозы 40 мг следует проводить только у пациентов с тяжелой формой гиперхолестеринемии и высоким риском сердечно-сосудистых осложнений (особенно у пациентов с наследственной гиперхолестеринемией), у которых при приеме дозы в 20 мг не была достигнута целевая концентрация холестерина. Такие пациенты должны находиться под врачебным наблюдением. Рекомендуется особенно тщательное наблюдение за пациентами, получающими препарат в дозе 40 мг. Не рекомендуется назначение дозы 40 мг пациентам, ранее не обращавшимся к врачу. После 2-4-х недель терапии и/или при повышении дозы препарата РОЗУКАРД® необходимо проводить контроль показателей липидного обмена (при необходимости требуется коррекция дозы). Пожилые пациенты У пациентов старше 65 лет коррекции дозы не требуется. Пациенты с печеночной недостаточностью У пациентов с печеночной недостаточностью со значениями но шкале Чайлд-Пью ниже 7 баллов коррекции дозы препарата РОЗУКАРД® не требуется. Лекарственный препарат РОЗУКАРД® противопоказан пациентам с заболеваниями печени в активной фазе, устойчивым повышением сывороточной активности "печеночных" трансаминаз (более чем в 3 раза по сравнению с верхней границей нормы (ВГН)) неясного генеза и пациентам с печеночной недостаточностью (степень тяжести от 7 до 9 баллов по шкале Чайлд-Пью) (см. раздел "Противопоказания"). Пациенты с почечной недостаточностью У пациентов с почечной недостаточностью легкой степени тяжести коррекции дозы не требуется. Рекомендуется начальная доза препарата РОЗУКАРД® - 5 мг в сутки. У пациентов с почечной недостаточностью тяжелой степени тяжести (КК менее 30 мл/мин) применение препарата РОЗУКАРД® противопоказано. У пациентов с почечной недостаточностью умеренной степени тяжести (КК 30 - 60 мл/мин) применение препарата РОЗУКАРД® в дозе 40 мг в сутки противопоказано (см. раздел "Противопоказания). Особые популяции. Этнические группы При изучении фармакокинетических параметров розувастатина было отмечено увеличение его системной концентрации у представителей монголоидной расы (см. раздел "Фармакокинетика"). Следует учитывать данный факт при назначении препарата РОЗУКАРД® пациентам монголоидной расы. При назначении доз 10 и 20 мг рекомендуемая начальная доза препарата РОЗУКАРД® для данной группы пациентов составляет 5 мг в сутки. Применение препарата РОЗУКАРД® в дозе 40 мг в сутки у представителей монголоидной расы противопоказано (см. раздел "Противопоказания). Генетический полиморфизм У носителей генотипов SLCO1B1 (ОАТР1В1) с.521СС и ABCG2 (BCRP) с.421АА отмечалось увеличение экспозиции (AUC) к розувастатину по сравнению с носителями генотипов SLC01B1 C.521TT и ABCG2 С.421СС. Для пациентов-носителей генотипов с.521СС или с.421АА рекомендуемая максимальная доза препарата РОЗУКАРД® составляет 20 мг в сутки (см. разделы "Фармакокинетика", "Особые указания" и "Взаимодействие с другими лекарственными средствами"). Пациенты с предрасположенностью к миопатии Применение препарата РОЗУКАРД® в дозе 40 мг в сутки противопоказано у пациентов с предрасположенностью к миопатии (см. раздел "Противопоказания). При назначении доз 10 мг и 20 мг в сутки рекомендуемая начальная доза препарата РОЗУКАРД® для данной группы пациентов составляет 5 мг в сутки. Сопутствующая терапия Розувастатин связывается с различными транспортными белками (в частности, с ОАТР1В1 и BCRP). При совместном применении препарата РОЗУКАРД® с лекарственными препаратами (такими как циклоспорин, некоторые ингибиторы протеазы ВИЧ, включая комбинацию ритонавира с атазанавиром, лопинавиром и/или типранавиром), повышающими концентрацию розувастатина в плазме крови за счет взаимодействия с транспортными белками, может повышаться риск развития миопатии (включая рабдомиолиз) (см. разделы "Особые указания" и "Взаимодействие с другими лекарственными средствами"). В таких случаях следует оценить возможность назначения альтернативной терапии или временного прекращения приема препарата РОЗУКАРД®. Если же применение указанных выше препаратов необходимо, следует ознакомиться с инструкцией по применению препаратов перед их назначением одновременно с препаратом РОЗУКАРД®, оценить соотношение пользы и риска сопутствующей терапии и рассмотреть возможность снижения дозы препарата РОЗУКАРД® (см. раздел "Взаимодействие с другими лекарственными средствами"). Побочные эффекты и передозировка Побочные эффекты: Побочные эффекты. наблюдаемые при приеме розувастатина, обычно выражены незначительно и проходят самостоятельно. Как и при применении других ингибиторов ГМГ-КоА-редуктазы, частота возникновения побочных эффектов носит, в основном, дозозависимый характер. Ниже представлен профиль нежелательных реакций для розувастатина, составленный на основании данных из клинических исследований и обширного пострегистрационного опыта. Частота побочных реакций определялась соответственно следующей градации (классификация Всемирной Организации Здравоохранения)- очень часто - более 1/10- часто - от более 1/100 до менее 1/10- нечасто - от более 1/1000 до менее 1/100- редко - от более 1 /10000 до менее 1 /1000- очень редко - от менее 1/10000. включая отдельные сообщения- частота неизвестна. Не представляется возможным установить частоту по имеющимся данным. Нарушения со стороны крови и лимфатической системы: редко - тромбоцитопения. Нарушения со стороны нервной системы: часто - головная боль, головокружение: очень редко - периферическая пейропатия. потеря или снижение памяти: частота неизвестна - нарушения сна, включая бессоницу и "кошмарные " сновидения. Нарушения психики: частота неизвестна - депрессия. Нарушения со стороны пищеварительной системы: часто - тошнота, запор, боль в животе: нечасто - рвота: редко - панкреатит: частота неизвестна - диарея. Нарушения со стороны печени и желчевыводящих путей: редко - дозозависимое транзиторное повышение активности аспартатаминотрансферазы (ACT) и аланинаминотрансферазы (АЛТ): очень редко - гепатит, желтуха. Нарушения со стороны дыхательной системы: частота неизвестна - кашель, диспноэ. Нарушения со стороны иммунной системы: редко - реакции гиперчувствительности, включая ангионевротический отек. Нарушения со стороны скелетно-мышечной и соединительной ткани: часто - миалгия: редко - миопатия (включая миозит), рабдомиолиз с острой почечной недостаточностью и без нее (у пациентов, получавших лечение в дозах >: 20 мг в сутки): очень редко - артралгия: частота неизвестна - иммуноопосредованная некротизирующая миопатия. Нарушения со стороны кожи и подкожных тканей: нечасто - кожный зуд, крапивница, кожная сыпь: частота неизвестна - синдром Стивенса-Джонсона. Нарушения со стороны почек и мочевыводящих путей: часто - протеинурия. Изменения количества белка в моче (от отсутствия или следовых количеств до + или больше) наблюдаются у менее 1 % пациентов, получающих дозу препарата 10-20 мг в сутки, и у приблизительно 3 % пациентов, получающих 40 мг в сутки. Протеинурия уменьшается в процессе терапии и не связана с возникновением заболевания почек или инфекцией мочевыводящих путей: очень редко - гематурия. Нарушения со стороны половых органов и молочной железы: очень редко - гинекомастия. Влияние на показатели лабораторных и инструментальных исследований: нечасто - дозозависимое повышение активности сывороточной креатинфосфокиназы (КФК). В большинстве случаев оно было незначительным, бессимптомным и временным. При повышении активности более чем в 5 раз по сравнению с ВГН терапия препаратом РОЗУКАРД® должна быть временно приостановлена. Повышение концентрации гликозилированного гемоглобина в плазме крови. Прочие: часто - астенический синдром, частота неизвестна - периферические отеки. При применении препарата РОЗУКАРД® отмечались изменения следующих лабораторных показателей: повышение концентрации глюкозы, билирубина, активности щелочной фосфатазы, гамма-глутамилтрапсферазы (ГГТ). О развитии следующих нежелательных явлений сообщалось во время применения некоторых статинов: - эректильная дисфункция: - единичные случаи иитерстициалыюго заболевания легких (особенно при длительном применении)- сахарный диабет 2 типа: частота зависит от наличия или отсутствия факторов риска (концентрация глюкозы в крови натощак 5.6-6.9 ммоль/л, индекс массы тела (ИМ Г) >: 30 кг/м 2, гипертриглицеридемия, артериальная гипертензия в анамнезе). Передозировка: При одновременном приеме нескольких суточных доз фармакокинетические параметры розувастатина не меняются. Лечение: специфического лечения нет, проводится симптоматическая терапия для поддержания функций жизненно важных органов и систем. Необходим контроль показателей функции печени и активности КФК. Гемодиализ неэффективен. Взаимодействие с другими ЛС: Влияние применения других препаратов на розувастатин. Ингибиторы транспортных белков: розувастатин связывается с некоторыми транспортными белками, в частности, с ОАТР1В1 и BCRP. Сопутствующее применение препаратов, которые являются ингибиторами этих транспортных белков, может сопровождаться увеличением концентрации розувастатина в плазме крови и повышенным риском развития миопатии (см. таблицу 3 и разделы "Способ применения и дозы" и "Особые указания"). Циклоспорин: при одновременном применении розувастатина и циклоспорина AUC розувастатина была в среднем в 7 раз выше значения, которое отмечалось у здоровых добровольцев (см. таблицу 3). Не влияет на плазменную концентрацию циклоспорина. Препарат РОЗУКАРД® противопоказан пациентам, принимающим циклоспорин (см. раздел "Противопоказания"). Ингибиторы протеазы вируса иммунодефицита человека (ВИЧ): несмотря на то, что точный механизм взаимодействия неизвестен, совместный приём ингибиторов протеазы ВИЧ может приводить к значительному увеличению экспозиции розувастатина (см. табл. 3). Фармакокинетическое исследование по одновременному применению 20 мг розувастатина с комбинированным препаратом, содержащим два ингибитора протеазы ВИЧ (400 мг лопинавира/100 мг ритонавира) у здоровых добровольцев приводило к приблизительно двукратному и пятикратному увеличению AUC(0-24) и Сmах розувастатина, соответственно. Поэтому одновременный прием препарата РОЗУКАРД® и ингибиторов протеазы ВИЧ не рекомендуется (см. разделы "Способ применения и дозы", "Особые указания", таблицу 3). Гемфиброзил и другие гиполипидемические средства: совместное применение розувастатина и гемфиброзила приводит к увеличению в 2 раза Сmах и AUC розувастатина (см. раздел "Особые указания"). Основываясь на данных по специфическому взаимодействию, не ожидается фармакокинетически значимого взаимодействия с фенофибратом, фармакодинамическое взаимодействие возможно. Гемфиброзил, фенофибрат, другие фибраты и липидснижающис дозы никотиновой кислоты (более 1 г в сутки) увеличивали риск возникновения миопатии при одновременном применении с ингибиторами ГМГ-КоА- редуктазы, возможно в связи с тем, что они могут вызывать миоиатию при применении в монотерапии (см. раздел "Особые указания"). При одновременном приёме препарата с гемфиброзилом, фибратами, никотиновой кислотой в липидснижающих дозах пациентам рекомендуется начальная доза препарата РОЗУКАРД® 5 мг, прием в дозе 40 мг противопоказан при совместном назначении с фибратами (см. разделы "Противопоказания", "Способ применения и дозы", "Особые указания"). Фузидовая кислота: конкретных исследований но изучению лекарственного взаимодействия фузидовой кислоты и розувастатина не проводилось, но были отмечены отдельные сообщения о случаях рабдомиолиза. Эзетимиб: одновременное применение препарата РОЗУКАРД® в дозе 10 мг и эзетимиба в дозе 10 мг сопровождалось увеличением AUC розувастатина у пациентов с гиперхолестсринемией (см. таблицу 3). Нельзя исключить увеличение риска возникновения побочных эффектов из-за фармакодинамического взаимодействия между препаратом РОЗУКАРД® и эзетимибом. Антациды: одновременное применение розувастатина и суспензий антацидов, содержащих магния и алюминия гидроксид. приводит к снижению плазменной концентрации розувастатина примерно на 50 %. Данный эффект выражен слабее, если антациды применяются через 2 часа после приема розувастатина. Клиническое значение подобного взаимодействия не изучалось. Эритромицин: одновременное применение розувастатина и эритромицина приводит к уменьшению AUC розувастатина на 20 % и Сmax розувастатина на 30 %. Подобное взаимодействие может возникать в результате усиления моторики кишечника, вызываемого приёмом эритромицина. Изоферменты системы цитохрома 1450: результаты исследований in vivo и in vitro показали, что розувастатин не является ни ингибитором, ни индуктором изоферментов системы цитохрома Р450. Кроме того, розувастатин является слабым субстратом для этих изоферментов. Поэтому не ожидается взаимодействия розувастатина с другими лекарственными средствами на уровне метаболизма с участием изоферментов системы цитохрома Р450. Не отмечено клинически значимого взаимодействия розувастатина с флуконазолом (ингибитором изоферментов CYP2C9 и CYP3A4) и кетоконазолом (ингибитором изоферментов CYP2A6 и CYP3A4). Взаимодействие с лекарственными средствами, которое требует коррекции дозы розувастатина (см. таблицу 3) Дозу препарата РОЗУКАРД® следует корректировать при необходимости его совместного применения с лекарственными средствами, увеличивающими экспозицию розувастатина. Если ожидается увеличение экспозиции в 2 раза и более, начальная доза препарата РОЗУКАРД® должна составлять 5 мг один раз в сутки. Также следует корректировать максимальную суточную дозу препарата РОЗУКАРД® так, чтобы ожидаемая экспозиция розувастатина не превышала таковую для дозы 40 мг, принимаемой без одновременного назначения лекарственных средств. взаимодействующих с розувастатином. Например, максимальная суточная доза препарата РОЗУКАРД® при одновременном применении с гемфиброзилом составляет 20 мг (увеличение экспозиции в 1.9 раза), с ритонавиром/атазанавиром - 10 мг (увеличение экспозиции в 3.1 раза). Таблица 3. Влияние сопутствующей терапии на экспозицию розувастатина (AUC, данные приведены в порядке убывания) - результаты опубликованных клинических исследований. ТАБЛИЦЪ Влияние применения розувастатина на другие препараты. Антагонисты витамина К: начало терапии или увеличение дозы препарата РОЗУКЛРД® у пациентов, получающих одновременно антагонисты витамина К (например, варфарин), может приводить к увеличению Международного Нормализованного Отношения (MHO). Отмена или снижение дозы препарата РОЗУКАРД® может приводить к уменьшению MHO. В таких случаях рекомендуется контроль M1IO. Пероральные контрацептивы/гормонозаместительная терапия: одновременное применение розувастатина и пероральных контрацептивов увеличивает AUС этинилэстрадиола и AUC норгестрела на 26 % и 34 %, соответственно. Такое увеличение плазменной концентрации должно учитываться при подборе дозы пероральных контрацептивов. Фармакокинетические данные по одновременному применению препарата РОЗУКАРД® и гормонозаместительной терапии отсутствуют, следовательно, нельзя исключить аналогичного эффекта и при применении данного сочетания. Однако подобная комбинация широко применялась во время проведения клинических исследований и хорошо переносилась пациентами. Другие лекарственные средства: не ожидается клинически значимого взаимодействия розувастатина с дигоксином. Фармакологическое действие и фармакокинетика Гиполипидемическос средство из группы статинов. Селективный конкурентный ингибитор 3-гидрокси-3-метилглутарил коэнзим А (ГМГ-КоА)-редуктазы - фермента, превращающего ГМГ-КоА в мевалонат. предшественник холестерина (ХС). Увеличивает число рецепторов липопротеинов низкой плотности (ЛИНИ) на поверхности гепатоцитов, что приводит к усилению захвата и катаболизма ЛИНИ, ингибированию синтеза липопротеинов очень низкой плотности (ЛПОНП). уменьшая общую концентрацию ЛИНИ и ЛПОНП. Снижает концентрации ХС-ЛПНП, холсстерина-нелипопротеинов высокой плотности (ХС-неЛПВП), ХС-ЛПОНП, общего ХС, триглицеридов (ТГ), ТГ-ЛПОНП, аполипопрогеина В (АпоВ), снижает соотношения ХС-ЛПНП/ХС-ЛПВП, общий ХС/ХС-ЛПВП, ХС-неЛПВП/ХС-ЛПВП, АпоВ/аполипонротеина А1 (АпоА1), повышает концентрации ХС-ЛПВП и АпоА1. Гиполипидемическое действие прямо пропорционально величине назначенной дозы. Терапевтический эффект появляется в течение 1 недели после начала терапии, через 2 недели достигает 90 % от максимального, к 4 неделе достигает максимума и после этого остается постоянным. Таблица 1. Дозозависимый эффект у пациентов с первичной гиперхолестеринемией (тип На и lib по классификации Фредриксоиа) (среднее скорректированное процентное изменение по сравнению с исходным значением). ТАБЛИЦЪ Клиническая эффективность Эффективен у взрослых пациентов с гиперхолестеринемией с или без гипертриглицеридемии (вне зависимости от расы, пола или возраста), в том числе у пациентов с сахарным диабетом и семейной гиперхолестеринемией. У 80 % пациентов с гиперхолестеринемией IIа и IIb типа (по классификации Фредриксона) со средней исходной концентрацией ХС-ЛПНП около 4,8 ммоль/л на фоне приема препарата в дозе 10 мг концентрация ХС-ЛПНП достигает значений менее 3 ммоль/л. У пациентов с гетерозиготной семейной гиперхолестеринемией, получающих розувастатин в дозе 20-80 мг в сутки, отмечается положительная динамика показателей липидного профиля. После титрования до суточной дозы 40 мг (12 недель терапии), отмечалось снижение концентрации ХС-ЛПНП на 53 %. У 33 % пациентов достигалась концентрация ХС-ЛПНП менее 3 ммоль/л. У пациентов с гомозиготной семейной гиперхолестеринемией, принимающих препарат в дозе 20 мг и 40 мг, среднее снижение концентрации ХС-ЛПНП составляло 22 %. У пациентов с гипертриглицеридемией с начальной концентрацией ТГ от 273 до 817 мг/дл, получавших розувастатин в дозе от 5 мг до 40 мг один раз в сутки в течение 6-ти недель, значительно снижалась концентрация ТГ в плазме крови (см. таблицу 2). Аддитивный эффект отмечается в комбинации с фенофибратом (в отношении снижения концентрации ТГ и с никотиновой кислотой в липидснижающих дозах (более 1 г/сутки) (в отношении снижения концентрации ХС- ЛПВП). (см. раздел "Особые указания"). В исследовании METEOR терапия розувастатином значительно замедляла скорость прогрессирования максимальной толщины комплекса "интима-медиа" (ТКИМ) для 12 сегментов сонной артерии по сравнению с плацебо. По сравнению с исходными значениями в группе розувастатина было отмечено уменьшение максимального значения ТКИМ на 0,0014 мм/год по сравнению с увеличением этого показателя на 0.0131 мм/год в группе плацебо. До настоящего времени прямой зависимости между уменьшением ТКИМ и снижением риска сердечнососудистых событий продемонстрировано не было. Результаты проведенного исследования JUPITER показали, что розувастатин существенно снижал риск развития сердечно-сосудистых осложнений со снижением относительного риска на 44 %. Эффективность терапии была отмечена через 6 первых месяцев применения препарата. Отмечено статистически значимое снижение на 48 % комбинированного критерия, включавшего смерть от сердечно-сосудистых причин, инсульт и инфаркт миокарда, уменьшение па 54 % возникновения фатального или нефатального инфаркта миокарда и на 48 % - фатального или нефатального инсульта. Общая смертность снизилась на 20 % в группе розувастатина. Профиль безопасности у пациентов, принимавших розувастатин в дозе 20 мг, был. в целом, схож с профилем безопасности в группе плацебо. Фармакокинетика: Всасывание. Максимальная концентрация (Сmaх) розувастатина в плазме крови достигается приблизительно через 5 ч после приема препарата. Абсолютная биодоступиость - примерно 20 %. Распределение. Проникает через плацентарный барьер. Розувастатин поглощается приемущественно печенью, которая является основным местом синтеза ХС и метаболизма ХС-ЛПНП. Объем распределения - 134 л. Связывание с белками плазмы крови (преимущественно с альбумином) составляет приблизительно 90 %. Метаболизм. Биотрансформируется в печени в небольшой степени (около 10 %). являясь непрофильным субстратом для изоферментов системы цитохрома Р450. Основным изоферментом, участвующим в метаболизме розувастатина, является изофермент CYP2C9. Изоферменты CYP2C19, CYP3A4 и CYP2D6 вовлечены в метаболизм в меньшей степени. Основными метаболитами розувастатина являются N- дисметилрозувастатин и лактоновые метаболиты. N-дисметилрозувастатин примерно на 50 % менее активен, чем розувастатин, лактоновые метаболиты фармакологически неактивны. Более 90 % фармакологической активности по ингибироваиию циркулирующей ГМГ-КоА-редуктазы обеспечивается розувастатином, остальное - его метаболитами. Выведение. Около 90 % дозы розувастатина выводится в неизмененном виде через кишечник, оставшаяся часть - почками. Период полувыведения (Т1/2) - примерно 19 ч, не изменяется при увеличении дозы препарата. Среднее значение геометрического плазменного клиренса составляет приблизительно 50 л/ч (коэффициент вариации 21.7 %). Как и в случае других ингибиторов ГМГ-КоА-редуктазы в процесс печеночного захвата препарата вовлечен специфический мембранный переносчик - полипептид, транспортирующий органический анион ОАТР1В1. выполняющий важную роль в его печеночной элиминации. Линейность. Системная экспозиция розувастатина увеличивается пропорционально дозе. Фармакокинетические параметры не изменяются при ежедневном приеме. Особые популяции пациентов. Почечная недостаточностьУ пациентов с почечной недостаточностью легкой и умеренной степени тяжести плазменные концентрации розувастатина или N-дисметилрозувастатина существенно не меняются. У пациентов с тяжелой почечной недостаточностью (клиренс креатинина (КК) менее 30 мл/мин) концентрация розувастатина в плазме крови выше в 3 раза, а N-дисметилрозувастатина - в 9 раз, чем у здоровых добровольцев. Концентрация розувастатина в плазме крови у пациентов, находящихся на гемодиализе, примерно на 50 % выше, чем у здоровых добровольцев. Печеночная недостаточностьУ пациентов с баллом 7 и ниже по шкале Чайлд-Пью не выявлено увеличения T1/2 розувастатина: у пациентов с баллами 8 и 9 по шкале Чайлд-Пью отмечено удлинение T1/2 в 2 раза. Опыт применения препарата у пациентов с более выраженными нарушениями функции печени отсутствует. Пол и возраст не оказывают клинически значимого влияния на фармакокинетику розувастатина. Этнические группы. Фармакокинетические параметры розувастатина зависят от расовой принадлежности: площадь под кривой "концентрация-время" (AUC) у представителей монголоидной расы (японцы, китайцы, филиппинцы, вьетнамцы и корейцы) в 2 раза выше, чем у представителей европеоидной расы. У индусов среднее значение AUC и Сmах увеличено в 1.3 раза. Генетический полиморфизм Ингибиторы ГМГ-КоА-редуктазы, в том числе розувастатин связываются с транспортными белками ОАТР1В1 (полипептид транспорта органических анионов, участвующий в захвате статинов гепатоцитами) и BCRP (эффлюксиый транспортер). У носителей генотипов SLC01B1 (ОАТР1В1) с.521СС и ABCG2 (BCRP) c.421АА отмечалось увеличение экспозиции (AUC) к розувастатину в 1,6 и 2,4 раза, соответственно, по сравнению с носителями генотипов SLC01B1 c.521ТТ и ABCG2 с.421СС. Особые указания Почечные эффекты У пациентов, получавших высокие дозы розувастатина (и основном 40 мг), наблюдалась канальцевая протеинурия, которая в большинстве случаев была транзиторной. Такая протеинурия не свидетельствовала об остром заболевании почек или прогрессировании заболевания почек. У пациентов, принимающих препарат в дозе 40 мг, рекомендуется контролировать показатели функции почек во время лечения. Эффекты на опорно-двигательный аппарат При применении розувастатина во всех дозах и. в особенности при приеме доз препарата, превышающих 20 мг. сообщалось о следующих воздействиях на опорно- двигательный аппарат: миалгия. миопатия, в редких случаях рабдомиолиз. Определение активности креатинфосфокиназы (КФК) Определение активности КФК не следует проводить после интенсивных физических нагрузок или при наличии других возможных причин увеличения активности КФК, что может привести к неверной интерпретации полученных результатов. В случае если исходная активность КФК существенно повышена (в 5 раз выше ВГН), через 5-7 дней следует провести повторное измерение. Не следует начинать терапию, если повторный тест подтверждает исходную активность КФК (более чем в 5 раз выше ВГН). До начала терапии При применении препарата РОЗУКАРД®, также как и при применении других ингибиторов ГМГ-КоА-редуктазы, у пациентов с имеющимися факторами риска миопатии/рабдомиолиза следует проявлять осторожность (см. раздел "С осторожностью"). Следует оценить соотношение риска и пользы и в случае необходимости терапии проводить клиническое наблюдение за пациентом во время лечения. Во время терапии Следует проинформировать пациента о необходимости немедленного сообщения врачу о случаях неожиданного появления мышечных болей, мышечной слабости или спазмах, особенно в сочетании с недомоганием и лихорадкой. У таких пациентов следует определять активность КФК. Терапия должна быть прекращена, если активность КФК значительно повышена (более чем в 5 раз выше ВГН) или если симптомы со стороны мышц резко выражены и вызывают ежедневный дискомфорт (даже если активность КФК увеличена менее чем в 5 раз по сравнению с ВГН). Если симптомы исчезают, и активность КФК возвращается к норме, следует рассмотреть вопрос о повторном назначении препарата РОЗУКАРД® или других ингибиторов ГМГ-КоА-редуктазы в меньших дозах при тщательном наблюдении за пациентом. Рутинный контроль активности КФК при отсутствии симптомов нецелесообразен. Отмечены очень редкие случаи иммуноопосредованной некротизирующей миопатии с клиническими проявлениями в виде стойкой слабости проксимальных мышц и повышения активности КФК в сыворотке крови во время лечения или при прекращении приема статинов, в том числе розувастатина. Может потребоваться проведение дополнительных исследований мышечной и нервной системы, серологических исследований, а также терапия иммунодепрессивными средствами. Не отмечено признаков увеличения воздействия на скелетную мускулатуру при приеме розувастатина и сопутствующей терапии. Однако сообщалось об увеличении числа случаев миозита и миопатии у пациентов, принимавших другие ингибиторы ГМГ-КоА-редуктазы в сочетании с производными фибриновой кислоты, включая гемфиброзил, циклоспорин, никотиновую кислоту в липидснижающих дозах (более I г/сутки), азольные противогрибковые средства, ингибиторы протеазы ВИЧ и макролидные антибиотики. Гемфиброзил увеличивает риск возникновения миопатии при совместном применении с некоторыми ингибиторами ГМГ- КоА-редуктазы, Таким образом, не рекомендуется одновременное применение препарата РОЗУКАРД® и гемфиброзила. Должно быть тщательно взвешено соотношение риска и возможной пользы при совместном применении препарата РОЗУКАРД® и фибратов или липидснижающих доз никотиновой кислоты. Противопоказан прием препарата РОЗУКАРД® в дозе 40 мг совместно с фибратами (см. разделы "Взаимодействие с другими лекарственными средствами", "Противопоказания"). Через 2-4 недели после начала лечения и/или при повышении дозы препарата РОЗУКАРД® необходим контроль показателей липидного обмена (при необходимости требуется коррекция дозы). Печень Рекомендуется проводить определение показателей функции печени до начала терапии и через 3 месяца после начала терапии. Прием препарата РОЗУКАРД® следует прекратить или уменьшить дозу препарата, если активность "печеночных" трансаминаз в плазме крови в 3 раза превышает ВГН. У пациентов с гиперхолестеринемией вследствие гипотиреоза или нефротического синдрома терапия основных заболеваний должна проводиться до начала лечения препаратом РОЗУКАРД®. Особые популяции. Этнические группы В ходе фармакокинетических исследований среди китайских и японских пациентов отмечено увеличение системной концентрации розувастатина по сравнению с показателями, полученными среди пациентов - европеоидов (см. разделы "Способ применения и дозы" и "Фармакокииетика"). Ингибиторы протеазы ВИЧ Не рекомендуется совместное применение препарата РОЗУКАРД® с ингибиторами протеазы ВИЧ (см. раздел "Взаимодействие с другими лекарственными средствами"). Лактоза Препарат РОЗУКАРД® не следует применять у пациентов с лактазной недостаточностью, непереносимостью галактозы и глюкозо-галактозной мальабсорбнией. Интерстициальное заболевание легких При применении некоторых статинов, особенно в течение длительного времени, сообщалось о единичных случаях интерстициального заболевания легких. Проявлениями заболевания могут являться одышка, непродуктивный кашель и ухудшение общего самочувствия (слабость, снижение массы тела и лихорадка). При подозрении на интерстициальное заболевание легких необходимо прекратить терапию препаратом РОЗУКАРД®. Сахарный диабет 2 типа Препараты класса статинов способны вызывать повышение концентрации глюкозы в крови. У некоторых пациентов с высоким риском развития сахарного диабета такие изменения могут приводить к его манифестации, что является показанием для назначения гипогликемической терапии. Однако снижение риска сосудистых заболеваний на фоне приема статинов превышает риск развития сахарного диабета, поэтому данный фактор не должен служить основанием для отмены лечения статинами. За пациентами группы риска (концентрация глюкозы в крови натощак 5.6- 6,9 ммоль/л, индекс массы тела (ИМТ) >: 30 кг/м 2, гипертриглицеридемия, артериальная гипертепзия в анамнезе) следует установить врачебное наблюдение и регулярно проводить контроль биохимических параметров. Влияние на способность управлять транспортными средствами: Следует соблюдать осторожность при управлении транспортными средствами и занятиях, требующих повышенной концентрации внимания и быстроты психомоторных реакций (во время терапии может возникать головокружение). Условия хранения и отпуска из аптек Условия хранения:При температуре не выше 25 °С в оригинальной упаковке. Хранить в недоступном для детей месте! Отпуск из аптек: По рецепту Регистрационные данные Торговое название Розукард® Международное непатентованное название:Розувастатин. Форма выпуска:таблетки покрытые пленочной оболочкой. Состав:Каждая таблетка покрытая пленочной оболочкой 10 мг содержит: активное вещество: розувастатин - 10,000 мг (в виде розувастатина кальция - 10,400 мг): вспомогательные вещества: ядро: лактозы моногидрат - 60,000 мг, целлюлоза микрокристаллическая - 45,400 мг, кроскармеллоза натрия - 1,200 мг, кремния диоксид коллоидный - 0,600 мг, магния стеарат - 2,400 мг: пленочная оболочка: гипромеллоза 2910/5 - 2,500 мг, макрогол 6000 - 0,400 мг, титана диоксид - 0,325 мг, тальк - 0,475 мг, краситель железа оксид красный - 0,013 мг. Каждая таблетка покрытая пленочной оболочкой 20 мг содержит: активное вещество: розувастатин - 20,000 мг (в виде розувастатина кальция - 20,800 мг): вспомогательные вещества: ядро: лактозы моногидрат - 120,000 мг, целлюлоза микрокристаллическая - 90,800 мг, кроскармеллоза натрия - 2,400 мг, кремния диоксид коллоидный - 1,200 мг, магния стеарат - 4,800 мг: пленочная оболочка: гипромеллоза 2910/5 - 5,000 мг, макрогол 6000 - 0,800 мг, титана диоксид - 0,650 мг, тальк - 0,950 мг, краситель железа оксид красный - 0,065 мг. Каждая таблетка покрытая пленочной оболочкой 40 мг содержит: активное вещество: розувастатин - 40,000 мг (в виде розувастатина кальция - 41,600 мг): вспомогательные вещества: ядро: лактозы моногидрат - 240,000 мг, целлюлоза микрокристаллическая - 181,600 мг, кроскармеллоза натрия - 4,800 мг, кремния диоксид коллоидный - 2,400 мг, магния стеарат - 9,600 мг: пленочная оболочка: гипромеллоза 2910/5 - 10,000 мг, макрогол 6000 - 1,600 мг, титана диоксид - 1,300 мг, тальк - 1,900 мг, краситель железа оксид красный - 0,200 мг. АТХ: Регистрация: Лекарственное средство ЛП-001704 Фармгруппа: гиполипидемическое средство - ГМГ-КоА редуктазы ингибитор. Дата регистрации: 18.05.2012/29.01.2016. Окончание регстрации: 2017-05-18 2017-05-18 . Описание:Таблетки 10 мг: продолговатые, двояковыпуклые таблетки с риской, покрытые плёночной оболочкой светло-розового цвета. Таблетки 20 мг: продолговатые, двояковыпуклые таблетки, покрытые плёночной оболочкой розового цвета. Таблетки 40 мг: продолговатые, двояковыпуклые таблетки, покрытые плёночной оболочкой темно-розового цвета. Упаковка:Таблетки, покрытые пленочной оболочкой, 10 мг, 20 мг, 40 мг. По 10 таблеток в блистере из А1/А1. По 3, 6 или 9 блистеров (для дозировок 10 мг, 20 мг) и по 3 или 9 блистеров (для дозировки 40 мг) помещены в картонную пачку вместе с инструкцией по применению. Срок годности:2 года. Препарат нельзя применять после истечения срока годности, указанного на упаковке. Владелец рег.удостоверения:Зентива к.с. Производитель:ZENTIVA, k.s. Представительство*:ЗЕНТИВА ФАРМА, ООО
849 Руб.Роксера таб. п/о 20мг №90
Показания Первичная гиперхолестеринемия по классификации Фредриксона (тип IIа, включая семейную гетерозиготную гиперхолестеринемню) или смешанная гиперхолестеринемия (тип IIb) - в качестве дополнения к диете, когда диета и другие немедикаментозные методы лечения (например, физическиеупражнения, снижение массы тела) оказываются недостаточными- Семейная гомозиготная гиперхолестеринемия - в качестве дополнения к диете и другой липидснижающей терапии (например, ЛПНП-аферез) или в случаях, когда подобная терапия недостаточно эффективна- Гипертриглицеридемия (тип IV по классификации Фредриксона) - в качестве дополнения к диете- Для замедления прогрессирования атеросклероза - в качестве дополнения к диете у пациентов, которым показана терапия для снижения плазменной концентрации общего ХС и ХС-ЛПНП- Первичная профилактика основных сердечно-сосудистых осложнений (инсульта, инфаркта миокарда, артериальной реваскуляризации) у взрослых пациентов без клинических признаков ишемической болезни сердца (ИБС), но с повышенным риском ее развития (возраст старше 50 лет для мужчин и старше 60 лет для женщин, повышенная плазменная концентрация С-реактивного белка (>: 2 мг/л) при наличии, как минимум одного из дополнительных факторов риска, таких как: артериальная гипертензия, низкая плазменная концентрация ХС-ЛПВП, курение, семейный анамнез раннего начала ИБС). Противопоказания Противопоказания При суточной дозе до 30 мг- Повышенная чувствительность к розувастатину или любому из компонентов препарата- заболевания печени в активной фазе (включая стойкое повышение активности "печеночных" трансаминаз и повышение активности "печеночных" трансаминаз в сыворотке крови более чем в 3 раза по сравнению с верхней границей нормы)- тяжелая почечная недостаточность (КК менее 30 мл/мин)- миопатия- одновременный прием циклоспорина- пациенты, предрасположенные к развитию миотоксических осложнений- беременность, период грудного вскармливания- применение у женщин детородного возраста, не пользующихся адекватными методами контрацепции- непереносимость лактозы, дефицит лактазы, синдром глюкозо-галактозной мальабсорбции- возраст до 18 лет. При суточной дозе 30 мг и более- Повышенная чувствительность к розувастатину или любому из компонентов препарата- заболевания печени в активной фазе (включая стойкое повышение активности "печеночных" трансаминаз и повышение активности "печеночных" трансаминаз в сыворотке крови более чем в 3 раза по сравнению с верхней границей нормы)- почечная недостаточность средней и тяжелой степени (КК менее 60 мл/мин)- миопатия- одновременное применение циклоспорина- пациенты, предрасположенные к развитию миотоксических осложнений- беременность, период грудного вскармливания- применение у женщин детородного возраста не пользующихся адекватными методами контрацепции- гипотиреоз- заболевания мышц в анамнезе (в том числе в семейном)- миотоксичность при применении других ингибиторов ГМГ-КоА-редуктазы или фибратов в анамнезе- чрезмерное употребление алкоголя- состояния, которые могут приводить к повышению концентрации розувастатина в плазме крови- одновременное применение фибратов- непереносимость лактозы, дефицит лактазы, синдром глюкозо-галактозной мальабсорбции- пациенты монголоидной расы- возраст до 18 лет. С осторожностью: При суточной дозе до 30 мг. Наличие риска развития миопатии/рабдомиолиза - почечная недостаточность, гипотиреоз, наследственные заболевания мышц в анамнезе (в том числе в семейном) и предшествующий анамнез мышечной токсичности при применении других ингибиторов ГМГ-КоА-редуктазы или фибратов: чрезмерное употребление алкоголя: возраст старше 65 лет: состояния, при которых отмечено повышение плазменной концентрации розувастатина: расовая принадлежность (монголоидная раса - японцы и китайцы): одновременное применение с фибратами: заболевания печени в анамнезе: сепсис: артериальная гипотензия: обширные хирургические вмешательства, травмы, тяжелые метаболические, эндокринные или электролитные нарушения или неконтролируемые судороги, одновременное применение с эзетимибом. При суточной дозе 30 мг и более. Почечная недостаточность легкой степени тяжести (КК более 60 мл/мин): возраст старше 65 лет: заболевания печени в анамнезе: сепсис: артериальная гипотензия: обширные хирургические вмешательства, травмы, тяжелые метаболические, эндокринные или электролитные нарушения или неконтролируемые судороги, одновременное применение с эзетимибом. Беременность Препарат Роксера® противопоказан при беременности и в период лактации. Женщины репродуктивного возраста должны применять адекватные методыконтрацепции. Поскольку холестерин и вещества, синтезируемые из холестерина важны для развития плода, потенциальный риск ингибирования ГМГ-КоА-редуктазы для плода превышает пользу от применения препарата при беременности.В случае наступления беременности в процессе терапии применение препарата должно быть немедленно прекращено. Данные в отношении выделения розувастатина с грудным молоком отсутствуют (известно, что другие ингибиторы ГМГ-КоА-редуктазы способные выделяться с грудным молоком), поэтому в период кормления грудью применение препарата необходимо прекратить. Применение и дозы Внутрь, таблетку не разжевывать и не измельчать, проглатывать целиком, запивая водой, возможен прием в любое время суток независимо от времени приема пищи. До начала терапии препаратом Роксера® пациент должен начать соблюдать стандартную гипохолестеринемическую диету и продолжать соблюдать ее во время лечения. Доза препарата должна подбираться индивидуально в зависимости от целей терапии и терапевтического ответа на лечение, принимая во внимание национальные рекомендации по целевым концентрациям липидов в плазме крови. Рекомендуемая начальная доза для пациентов, начинающих принимать препарат, или для пациентов, переведенных с приема других ингибиторов ГМГ-КоА-редуктазы, должна составлять 5 или 10 мг препарата Роксера® 1 раз в сутки. При одновременном применении препарата с гемфиброзилом, фибратами, никотиновой кислотой в липидснижающих дозах (более 1 г/сут) пациентам рекомендуется начальная доза препарата 5 мг/сут. При выборе начальной дозы следует руководствоваться индивидуальной концентрацией холестерина в плазме крови и принимать во внимание возможный риск развития сердечно-сосудистых осложнений, необходимо также учитывать потенциальный риск развития побочных эффектов. В случае необходимости, доза может быть увеличена через 4 недели. В связи с возможным развитием побочных эффектов при применении дозы 40 мг/сут, по сравнению с более низкими дозами препарата, повышение дозы до максимальной 40 мг/сут следует рассматривать только у пациентов с тяжелой степенью гиперхолестеринемии и с высоким риском развития сердечно-сосудистых осложнений (особенно у пациентов с семейной гиперхолестеринемией), у которых не был достигнут желаемый результат терапии при применении дозы 20 мг/сут, и которые будут находиться под наблюдением врача. Рекомендуется особенно тщательное наблюдение за пациентами, получающими препарат в дозе 40 мг/сут. Не рекомендуется применение дозы 40 мг/сут у пациентов, ранее не обращавшихся к врачу. После 2-4 недель терапии и/или при повышении дозы препарата Роксера® необходим контроль показателей липидного обмена (при необходимости требуется коррекция дозы). Пациенты с почечной недостаточностью У пациентов с почечной недостаточностью легкой или умеренной степени тяжести коррекции дозы не требуется. У пациентов с тяжелой почечной недостаточностью (КК менее 30 мл/мин) применение препарата Роксера® противопоказано. Применение препарата Роксера® в дозе более 30 мг/сут пациентам с почечной недостаточностью умеренной и тяжелой степени (КК менее 60 мл/мин) противопоказано. Пациентам с почечной недостаточностью умеренной степени рекомендуемая начальная доза препарата Роксера® составляет 5 мг/сут. Пациенты с печеночной недостаточностью Препарат Роксера® противопоказан пациентам с заболеваниями печени в активной фазе. Применение у пациентов пожилого возраста Не требуется коррекции дозы. Этнические группыУ пациентов монголоидной расы отмечено увеличение системной экспозиции розувастатина. Для пациентов монголоидной расы рекомендуемая начальная доза препарата Роксера® составляет 5 мг/сут, применение препарата Роксера® дозе 40 мг противопоказано. Генетический полиморфизмУ носителей генотипов SLC01B1 (ОАТР1В1) с.521СС и ABCG2 (BCRP) с.421АА отмечалось увеличение экспозиции (AUC) розувастатина по сравнению с носителями генотипов SLCO1B1 с.521ТТ и ABCG2 с.421СС. Для пациентов-носителей генотипов с.521СС или с.421АА рекомендуемая максимальная доза препарата Роксера® составляет 20 мг 1 раз в сутки. Пациенты, предрасположенные к миотоксическим осложнениям Применение препарата Роксера® в дозе 40 мг пациентам, предрасположенным к развитию миотоксических осложнений противопоказано. При необходимости применения доз К) и 20 мг/сут рекомендуемая начальная доза для данной группы пациентов составляет 5 мг/сут. Сопутствующая терапия. Розувастатин связывается с различными транспортными белками (в частности, с ОАТР1В1 и BCRP). При одновременном применении препарата Роксера с лекарственными препаратами (такими как циклоспорин, некоторые ингибиторы протеазы вируса иммунодефицита человека (ВИЧ), включая комбинацию ритонавира с атазапавиром, лопипавиром и/или типранавиром), повышающими концентрацию розувастатина в плазме крови за счет взаимодействия с траспортными белками, может повышаться риск развития миопатии (включая рабдомиолиз). Следует ознакомиться с инструкцией по применению вышеуказанных препаратов перед их назначением одновременно с препаратом Роксера®. В таких случаях следует оценить возможность применения альтернативной терапии или временного прекращения приема препарата Роксера®. При необходимости применения указанных выше препаратов, следует оценить соотношение пользы и риска сопутствующей терапии препаратом Роксера® и рассмотреть возможность снижения его дозы. Побочные эффекты и передозировка Побочные эффекты: Побочные эффекты, наблюдаемые при приеме препарата Роксера®, обычно выражены незначительно и проходят самостоятельно. Как и при применении других ингибиторов ГМГ-КоА-редуктазы, частота возникновения Абсорбция и распределение Максимальная концентрация розувастатина в плазме крови достигается приблизительно через 5 ч после приема внутрь. Абсолютная биодоступность составляет примерно 20 %. Метаболизируется преимущественно печенью, которая является основным органом, синтезирующим холестерин и метаболизирующим ХС-ЛПНП. Объем распределения розувастатина составляет примерно 134 л. Приблизительно 90 % розувастатина связывается с белками плазмы крови, в основном с альбумином. Метаболизм Подвергается ограниченному метаболизму (около 10 %). Розувастатин является побочных эффектов носит, в основном, дозозависимый характер. Классификация частоты развития побочных эффектов Всемирной организации здравоохранения (ВОЗ): очень часто >: 1/10 часто от >: 1/100 до <: 1/10 нечасто от >: 1/1000 до <: 1/100 редко от >: 1/10000 до <: 1/1000 очень редко <: 1/10000 частота неизвестна не может быть оценена на основе имеющихся данных. Нарушения со стороны крови и лимфатической системы: частота неизвестна: тромбоцитопения. Нарушения со стороны иммунной системы: редко: реакции повышенной чувствительности, включая ангионеврогический отёк. Нарушения со стороны эндокринной системы: часто: сахарный диабет 2-го типа. Нарушения со стороны нервной системы: часто: головная боль, головокружение: очень редко: потеря или снижение памяти: частота неизвестна: периферическая нейропатия. Нарушения со стороны дыхательной системы, органов грудной клетки и средостения:частота неизвестна: кашель, одышка. Нарушения со стороны пищеварительной системы:часто: запор, тошнота, боль в животе:редко: панкреатит:очень редко: желтуха, гепатит:частота неизвестна: диарея. При применении розувастатина наблюдается дозозависимое повышение активности "печеночных" трансамнназ в плазме крови у незначительного числа пациентов. В большинстве случае оно незначительно, бессимптомно и временно. Нарушения со стороны кожи и подкожных тканей:нечасто: кожный зуд, кожная сыпь, крапивница:частота неизвестна: синдром Стивенса-Джонсона. Нарушения со стороны скелетно- мышечной и соединительной ткани:часто: миалгия:редко: миопатия (включая миозит), рабдомиолиз (с острой почечной недостаточностью или без нее):очень редко: артралгия:частота неизвестна: иммуноопосредованная некротизирующая миопатия. Дозозависимое повышение активности креатинфосфокиназы (КФК) в плазме крови наблюдается у небольшого числа пациентов, принимавших розувастатин. В большинстве случаев оно является незначительным, бессимптомным и временным. В случае повышения активности КФК в плазме крови более чем в 5 раз выше верхней границы нормы терапию следует приостановить. Нарушения со стороны почек и мочевыводящих путей:У пациентов, получающих терапию розувастатином, может выявляться протеинурия. Изменение количества белка в моче (от отсутствия или следовых количеств до + или больше) наблюдается менее чем у 1 % пациентов, получающих 10-20 мг препарата, и у приблизительно 3 % пациентов, получающих 40 мг препарата. Незначительное изменение количества белка в моче отмечалось при приеме дозы 20 мг. В большинстве случаев протеинурия уменьшается или исчезает в процессе терапии и не означает возникновенияострого или прогрессирования существующего заболевания почек. Очень редко: гематурия. Нарушения со стороны половых органов и молочной железы:частота неизвестна: гинекомастия. Общие расстройства и нарушения в месте введения: часто: астенический синдром: частота неизвестна: периферические отеки. Лабораторные показатели:При применении розувастатина также наблюдались следующие изменения лабораторных показателей: гипергликемия, повышение концентрации билирубина в плазме крови, активности гамма-глутамилтраиспептидазы, щелочной фосфатазы в плазме крови, изменение сывороточной концентрации гормонов щитовидной железы. При применении некоторых ингибиторов ГМГ-КоА-редуктазы (статинов) сообщалось о следующих побочных эффектах: депрессия, нарушения сна, включая бессонницу и "кошмарные" сновидения, сексуальная дисфункция, повышение концентрации гликозилированного гемоглобина. Сообщалось о единичных случаях интерстициального заболевания легких, особенно при длительном применении препаратов (см. раздел "Особые указания"). Передозировка: Клиническая картина передозировки не описана. При единовременном приеме нескольких суточных доз препарата фармакокинетические параметры розувастатина не изменяются. Лечение передозировки - симптоматическое, необходим контроль функции печени и активности КФК: специфического антидота не существует, гемодиализ неэффективен. Взаимодействие с другими ЛС: Влияние применения других препаратов на розувастатин Ингибиторы транспортных белков Розувастатин является субстратом для некоторых транспортных белков, в частности, ОАТР1В1 и BCRP. Одновременное применение препаратов, которые являются ингибиторами этих транспортных белков,может сопровождаться увеличением концентрации розувастатина в плазме крови и повышенным риском развития миопатии (см. таблицу 1, разделы "Способ применения и дозы", "Особые указания"). Циклоспорин При одновременном применении розувастатина и циклоспорина, AUC розувастатина в среднем в 7 раз выше значения, которое отмечается у здоровых добровольцев (см. таблицу 1). Одновременноеприменение с розувастатином не влияет на концентрацию циклоспорина в плазме крови. Применение розувастатина противопоказано пациентам, принимающим циклоспорин (см. раздел "Противопоказания"). Ингибиторы протеазы ВИЧ Одновременное применение ингибиторов протеазы ВИЧ может значительно повышать экспозицию розувастатина (см. таблицу 1). Одновременное применение 20 мг розувастатина и комбинации двух ингибиторов протеазы ВИЧ (400 мг лопинавира/100 мгритонавира) сопровождается повышением равновесных AUC(0-24) и Сmах розувастатина в 2 и 5 раз, соответственно. Поэтому одновременный прием розувастатина и ингибиторов протеазы ВИЧ не рекомендуется (см. раздел "Способ применения и дозы", таблицу 1). Гемфиброзил и другие гиполипидемические средства Одновременное применение розувастатина и гемфиброзила приводит к увеличению Сmax и AUC розувастатина в плазме крови в 2 раза (см. раздел "Особые указания"). Основываясь на данных по специфическому взаимодействию, не ожидается фармакокинетически значимого взаимодействия с фенофибратом, возможно фармакодинамическое взаимодействие. Гемфиброзил, фенофибрат, другие фибраты и липидснижающие дозы никотиновой кислоты (дозы большие или эквивалентные 1 г/сут) увеличивали риск возникновения миопатии при одновременном применении с ингибиторами ГМГ-КоА-редуктазы, возможно в связи с тем, что они могут вызывать миопатию при применении в монотерапии (см. раздел "Особые указания"). Одновременное применение фибратов и розувастатина в суточной дозе 30 мг противопоказано. У таких пациентов терапия должна начинаться с дозы 5 мг/сут (см. разделы "Противопоказания", "Способ применения и дозы", "Особые указания"). Эзетимиб Одновременное применение розувастатина в дозе 10 мг и эзетимиба в дозе 10 мг сопровождалось увеличением AUC розувастатина у пациентов с гиперхолестеринемией (см. таблицу 1). Нельзя исключить фармакодинамическое взаимодействие между розувастатином и эзегимибом, проявляющееся увеличением риска развития нежелательных реакций. Антациды Одновременное применение розувастатина и антацидов, содержащих алюминия и магния гидроксид, приводит к снижению плазменной концентрации розувастатина примерно на 50 %. Данный эффект выражен слабее, если антациды применяются через 2 ч после приема розувастатина. Клиническое значение подобного взаимодействия не изучалось. Эритромицин Одновременное применение розувастатина и эритромицина приводит к уменьшению AUC(o-t) розувастатина на 20 % и его Сmах на 30 %. Подобное взаимодействие может возникать в результате усиления моторики кишечника, вызываемого применением эритромицина. Изоферменты системы цитохрома Р450 Результаты исследований in vivo и in vitro показали, что розувастатин не является ни ингибитором, ни индуктором изоферментов системы цитохрома Р450. Кроме того, розувастатин является слабым субстратом для этой системы изоферментов. Поэтому неожидается взаимодействия розувастатина с другими лекарственными средствами (ЛС) на уровне метаболизма с участием изоферментов системы цитохрома Р450. Клинически значимого взаимодействия между розувастатином и флуконазолом (ингибитором изоферментов CYP2C9 и CYP3A4) и кетоконазолом (ингибитором изоферментов CYP2A6 и CYP3A4) не отмечено. Фузидовая кислота Исследований по изучению взаимодействия розувастатина и фузидовой кислоты не проводилось. Как и при приеме других статинов, были полученыпостмаркетинговые сообщения о случаях рабдомиолиза при одновременномприменении розувастатина и фузидовой кислоты. Необходимо пристально наблюдать за пациентами. При необходимости, возможно временное прекращение приема розувастатина. Взаимодействие с лекарственными средствами, которое требует коррекции дозы розувастатина (см. таблицу 1) Дозу препарата Роксера® следует корректировать при необходимости его одновременного применения с ЛC, увеличивающими экспозицию розувастатина. Если ожидается увеличение экспозиции в 2 раза и более, начальная доза препарата Роксера® должна составлять 5 мг 1 раз в сутки. Также следует корректировать максимальную суточную дозу препарата Роксера®, чтобы ожидаемая экспозиция розувастатина не превышала таковую для дозы 40 мг, принимаемой без одновременного назначения ЛC, взаимодействующих с розувастатином. Например, максимальная суточная доза препарата Роксера® при одновременном применении с гемфиброзилом составляет 20 мг (увеличение экспозиции в 1,9 раза), с ритонавиром/атазанавиром - 10 мг (увеличение экспозиции в 3,1 раза). Таблица 1. Влияние сопутствующей терапии на экспозицию розувастатина (AUC, данные приведены в порядке убывания) - результаты опубликованных клинических исследований ТАБЛИЦЪ Влияние применения розувастатина на другие препараты Антагонисты витамина К Как и в случае других ингибиторов ГМГ- КоА-редукгазы, начало терапии розувастатином или увеличение его дозы у пациентов, принимающих одновременно антагонисты витамина К (например, варфарин), может приводить к увеличению Международного нормализованного отношения (MHO). Отмена розувастатина или снижение его дозы может приводить к уменьшению MHO. В таких случаях рекомендуется мониторинг MHO. Контрацептивы для приема внутрь/заместительная гормональная терапия (ЗГТ) Одновременное применение розувастатина и контрацептивов для приема внутрь увеличивает AUC этинилэстрадиола и норгестрела на 26 % и 34 %, соответственно. Такое увеличение концентрации в плазме крови должно учитываться при подборе дозы гормональных контрацептивов. Фармакокинетические данные по одновременному применению розувастатина и ЗГТ отсутствуют, следовательно, нельзя исключить аналогичного эффекта и при применении данной комбинации. Однако подобная комбинация широко применялась во время проведения клинических исследований и хорошо переносилась пациентами. Другие лекарственные средства Клинически значимого взаимодействия розувастатина с дигоксином не ожидается. Фармакологическое действие и фармакокинетика Препарат Роксера® - гиполипидемическое средство. Действующее вещество препарата -розувастатин, является селективным, конкурентным ингибитором ГМГ-КоА редуктазы, фермента, превращающего З-гидрокси-З-метилглутарилкофермент А в мевалоновую кислоту - предшественник холестерина. Основной мишенью действия розувастатина является печень, где происходит синтез холестерина (ХС) и катаболизм липопротеинов низкой плотности (ЛПНП). Увеличивает число печеночных рецепторов к ЛПНП на поверхности клеток, повышая захват и катаболизм ЛПНП, что в свою очередь приводит к ингибированию синтеза липопротеинов очень низкой плотности (ЛПОНП), уменьшая тем самым общее количество ЛПНП и ЛПОНП. Розувастатин снижает повышенные плазменные концентрации холестерина ЛПНП (ХС-ЛПНП), общего холестерина, триглицеридов (ТГ), повышает концентрацию холестерина липопротеинов высокой плотности (ХС-ЛПВП). Он также снижает концентрацию аполипопротеина В (АпоВ), ХС-неЛПВП, ХС-ЛПОНП, ТГ-ЛПОНП и увеличивает концентрацию аполипопротеина A-I в плазме крови. Розувастатин снижает соотношение ХС-ЛПНП/ХС-ЛПВП, общий ХС/ХС-ЛПВП и ХС-неЛПВП/ ХС-ЛПВП и соотношение АпоВ/АпоА-1. Терапевтический эффект развивается в течение одной недели после начала терапии, через 2 недели лечения достигает 90 % от максимально возможного эффекта. Максимальный терапевтический эффект обычно достигается к 4 неделе терапии и поддерживается при дальнейшем регулярном приёме препарата. Клиническая эффективность. Розувастатин эффективен у взрослых пациентов с гиперхолестеринемией с сопутствующей гипертриглицеридемией или без нее, вне зависимости от расовой принадлежности, пола или возраста, в том числе у пациентов с сахарным диабетом или семейной гиперхолестеринемией.У 80 % пациентов с гиперхолестеринемией Па и lib типа по Фредриксону (средняя исходная концентрация ХС-ЛПНП примерно 4,8 ммоль/л) на фоне приема розувастатина в дозе 10 мг концентрация ХС-ЛПНП достигает значений менее ммоль/л. У пациентов с гомозиготной семейной гиперхолестеринемией розувастатин применялся в дозах 20-40 мг, среднее снижение составило 22 %. Аддитивный эффект отмечается в комбинации с фенофибратом в отношении содержания ТГ и с никотиновой кислотой в липидснижающих дозах в отношении концентрации ХС-ЛПВП. Фармакокинетика: Абсорбция и распределение Максимальная концентрация розувастатина в плазме крови достигается приблизительно через 5 ч после приема внутрь. Абсолютная биодоступность составляет примерно 20 %. Метаболизируется преимущественно печенью, которая является основным органом, синтезирующим холестерин и метаболизирующим ХС-ЛПНП. Объем распределения розувастатина составляет примерно 134 л. Приблизительно 90 % розувастатина связывается с белками плазмы крови, в основном с альбумином. Метаболизм Подвергается ограниченному метаболизму (около 10 %). Розувастатин является неспецифическим субстратом цитохрома Р450. Основным изоферментом, участвующим в метаболизме розувастатина, является изоферменг CYP2C9. Изоферменты CYP2C19, CYP3A4, CYP2D6 вовлечены в метаболизм в меньшей степени. Основными выявленными метаболитами являются N-десметилрозувастатин и лактоновые метаболиты. N-десметилрозувастатин примерно на 50 % менее активен, чем розувастатин, лактоновые метаболиты фармакологически неактивны. Более 90 % фармакологической активности по ингибированию плазменной ГМГ-КоА-редуктазы обеспечивается розувастатином, остальное - его метаболитами. Выведение Около 90 % дозы розувастатина выводится в неизмененном виде через кишечник (включая абсорбированный и неабсорбированный розувастатин). Оставшаяся часть выводится почками. Период полувыведения (Т1/2) из плазмы крови составляет примерно 19 ч (не изменяется при увеличении дозы препарата). Средний геометрический плазменный клиренс - 50 л/ч (коэффициент вариации - 21,7 %). Как и в случае других ингибиторов ГМГ-КоА-редуктазы, в процесс "печеночного" захвата розувастатина вовлечен мембранный переносчик холестерина, выполняющий важную роль в "печеночной" элиминации розувастатина. Линейность Системная экспозиция розувастатина увеличивается пропорционально дозе. Фармакокинетические параметры не изменяются при ежедневном приеме. Особые группы пациентов Возраст и пол Пол и возраст не оказывают клинически значимого влияния на фармакокинетику розувастатина. Этнические группы. Фармакокинетические исследования показали приблизительно двукратное увеличение медианы AUC (площади под кривой "концентрация-время") и максимальной концентрации (Сmах) в плазме крови розувастатина у пациентов монголоидной расы (японцев, китайцев, филиппинцев, вьетнамцев и корейцев) по сравнению с пациентами европеоидной расы: у индийцев показано увеличение медианы AUC и Сmах в 1,3 раза. Фармакокинетический анализ не выявил клинически значимых различий в фармакокинетике среди пациентов европеоидной и негроидной рас. Почечная недостаточностьУ пациентов с почечной недостаточностью легкой или умеренной степени тяжести величина плазменной концентрации розувастатина или N-десметилрозувастатина существенно не меняется. У пациентов с тяжелой почечной недостаточностью (клиренс креатинина (КК) менее 30 мл/мин) концентрация розувастатина в плазме крови в 3 раза выше, а концентрация N-десмегилрозувастатина в 9 раз выше, чем у здоровых добровольцев. Концентрация розувастатина в плазме крови у пациентов, находящихся на гемодиализе, примерно на 50 % выше, чем у здоровых добровольцев. Печеночная недостаточность У пациентов с печеночной недостаточностью 7 баллов и ниже по шкале Чайлд-Пью не выявлено увеличения системной экспозиции розувастатина. У двух пациентов с печеночной недостаточностью 8-9 баллов по шкале Чайлд-Пью отмечено увеличение системной экспозиции, по крайней мере, в 2 раза. Опыт применения розувастатина у пациентов с печеночной недостаточностью выше 9 баллов по шкале Чайлд-Пью отсутствует. Генетический полиморфизм Ингибиторы ГМГ-КоА-редуктазы, в том числе розувастатин, связываются с транспортными белками ОАТР1В1 (полипептид транспорта органических анионов, участвующий в захвате статинов гепатоцитами) и BCRP (эффлюксный транспортер). У носителей генотипов SLCO1B1 (ОАТР1В1) с.521СС и ABCG2 (BCRP) c.421АА отмечалось увеличение экспозиции (AUC) розувастатина в 1,6 и 2,4 раза, соответственно, по сравнению с носителями генотипов SLCO1B1 c.521ТТ и ABCG2 с.421СС. Особые указания Нарушение функции почек У пациентов, получавших высокие дозы розувастатина (в частности 40 мг/сут), наблюдалась канальцевая протеинурия, которая выявлялась при помощи тест- полосок и в большинстве случаев была периодической или кратковременной. Такая протеинурия не свидетельствует об остром или прогрессировании сопутствующего заболевания почек. Частота серьезных нарушений функции почек, отмеченная при постмаркетинговом изучении розувастатина, выше при приёме дозы 40 мг/сут. У пациентов, принимающих препарат Роксера® в дозе 30 или 40 мг/сут, рекомендуется контролировать показатели функции почек во время лечения (не реже чем 1 раз в 3 месяца). Влияние на опорно-двигательный аппарат При применении розувастатина во всех дозах, но в особенности в дозах, превышающих 20 мг/сут, сообщалось о следующих воздействиях на опорно- двигательный аппарат: миалгия, миопатия, в редких случаях рабдомиолиз. Отмечены очень редкие случаи рабдомиолиза при одновременном применении ингибиторов ГМГ-КоА-редуктазы и эзетимиба. Такая комбинация должна применяться с осторожностью, так как нельзя исключить фармакодинамического взаимодействия. Как и в случае других ингибиторов ГМГ-КоА-редуктазы, частота рабдомиолиза при постмаркетинговом применении препарата Роксера® выше при применении дозы 40 мг/сут. Определение активности КФК Активность КФК нельзя определять после интенсивных физических нагрузок и при наличии других возможных причин повышения ее активности: это может привести к неверной интерпретации полученных результатов. В случае если исходная активность КФК существенно превышена (в 5 раз выше верхней границы нормы), через 5-7 дней следует провести повторный анализ. Нельзя начинать терапию, если результаты повторного анализа подтверждают исходную высокую активность КФК (более чем 5-кратное превышение верхней границы нормы). Перед началом терапии В зависимости от суточной дозы препарат Роксера® должен назначаться с осторожностью пациентам с имеющимися факторами риска миопатии/рабдомиолиза или применение препарата противопоказано (см. разделы "Противопоказания" и "С осторожностью"). К таким факторам относятся: - нарушение функции почек: - гипотиреоз: - заболевания мышц в анамнезе (в том числе в семейном): - миотоксические явления при приёме других ингибиторов ГМГ-КоА-редуктазы или фибратов в анамнезе: - чрезмерное употребление алкоголя: - возраст старше 65 лет: - состояния, при которых может повышаться концентрация розувастатина в плазме крови- одновременное применение фибратов. У таких пациентов необходимо оценить риск и возможную пользу терапии. Также рекомендуется проводить клинический мониторинг. Если исходная активность КФК выше более чем в 5 раз по сравнению с верхней границей нормы, терапию препаратом Роксера® начинать нельзя. В период терапии препаратом Следует проинформировать пациента о необходимости немедленного сообщения врачу в случае неожиданного появления мышечных болей, мышечной слабости или спазмов, особенно в сочетании с недомоганием и лихорадкой. У таких пациентов следует определять активность КФК. Терапия должна быть прекращена, если активность КФК значительно увеличена (более чем в 5 раз по сравнению с верхней границей нормы) или если симптомы со стороны мышц резко выражены и вызывают ежедневный дискомфорт (даже, если активность КФК не более чем в 5 раз превышает верхнюю границу нормы). Если симптомы исчезают и активность КФК возвращается к норме, следует рассмотреть вопрос о возобновлении применения препарата Роксера® или других ингибиторов ГМГ-КоА-редуктазы в меньших дозах при тщательном медицинском наблюдении. Контроль активности КФК при отсутствии симптомов нецелесообразен. Отмечены очень редкие случаи иммуноопосредованной некротизирующей миопатии с клиническими проявлениями в виде стойкой слабости проксимальных мышц и повышения активности КФК в сыворотке крови во время терапии или при прекращении применения ингибиторов ГМГ-КоА-редуктазы, в том числе розувастатина. Может потребоваться проведение дополнительных исследований мышечной и нервной системы, серологических исследований, а также терапия иммунодепрессивными средствами. Признаков увеличения воздействия на скелетную мускулатуру при приеме розувастатина и сопутствующей терапии не отмечено. Однако сообщалось об увеличении числа случаев миозита и миопатии у пациентов, принимавших другие ингибиторы ГМГ-КоА-редуктазы в сочетании с производными фиброевой кислоты (например, гемфиброзил), циклоспорином, никотиновой кислотой в липидснижающих дозах (более 1 г/сутки), противогрибковыми средствами производными азола, ингибиторами протеазы ВИЧ и макролидными антибиотиками. При одновременном применении с некоторыми ингибиторами ГМГ-КоА- редуктазы гемфиброзил увеличивает риск развития миопатии. Таким образом, одновременное применение препарата Роксера® и гемфиброзила не рекомендуется. Преимущества дальнейшего изменения плазменной концентрации липидов при комбинированном применении препарата Роксера® с фибратами или никотиновой кислотой в липидснижающих дозах должны быть тщательно взвешены с учётом возможного риска. Препарат Роксера® в дозе 30 мг/сут противопоказан для комбинированной терапии с фибратами. В связи с увеличением риска рабдомиолиза препарат Роксера® не следует применять пациентам с острыми состояниями, которые могут привести к миопатии или состояниям, предрасполагающим к развитию почечной недостаточности (например, сепсис, артериальная гипотензия, обширные хирургические вмешательства, травмы, тяжелые метаболические, эндокринные и электролитные нарушения или неконтролируемые судороги). Печень В зависимости от суточной дозы препарат Роксера® должен с осторожностью применяться у пациентов с чрезмерным употреблением алкоголя и/или имеющих в анамнезе заболевания печени или его применение противопоказано (см. разделы "Противопоказания" и "С осторожностью"). Рекомендуется проводить определение функциональных проб печени до начала терапии и через 3 месяца после ее начала. Применение препарата Роксера® следует прекратить или уменьшить дозу препарата, если активность "печеночных" трансаминаз в сыворотке крови в 3 раза превышает верхнюю границу нормы.У пациентов с гиперхолестеринемией вследствие гипотиреоза или нефротического синдрома до начала лечения препаратом Роксера® должна проводиться терапия основных заболеваний. Этнические особенностиВ ходе фармакокинетических исследований у представителей монголоидной расы по сравнению с представителями европеоидной расы отмечено увеличение плазменной концентрации розувастатина. Препарат Роксера® содержит лактозу, в связи с чем его не следует применять пациентам с непереносимостью лактозы, дефицитом лактазы, синдромом глюкозо- галактозной мальабсорбции. Интерстициалъное заболевание легких. При применении некоторых ингибиторов ГМГ-КоА-редуктазы, особенно в течение длительного времени, сообщалось о единичных случаях интерстициального заболевания легких. Проявлениямизаболевания могут являться одышка, непродуктивный кашель и ухудшение общего самочувствия (слабость, снижение веса и лихорадка). При подозрении на интерстициальное заболевание легких следует прекратить терапию ингибиторами ГМГ-КоА- редуктазы. Сахарный диабет 2-го типаУ пациентов с концентрацией глюкозы от 5,6 до 6,9 ммоль/л терапия розувастатином ассоциировалась с повышенным риском развития сахарного диабета 2-го типа. Влияние на способность управлять транспортными средствами: исследования по изучению влияния препарата Роксера® на способность управлять транспортными средствами и работу с механизмами не проводились. Тем не менее, учитывая возможность развития головокружения и др. побочных эффектов, необходимо соблюдать осторожность при управлении транспортными средствами и другими механизмами, требующими повышенной концентрации внимания и быстроты психомоторных реакций. Условия хранения и отпуска из аптек Условия хранения:При температуре не выше 25 °С, в оригинальной упаковке. Хранить в недоступном для детей месте. Отпуск из аптек: По рецепту Регистрационные данные Торговое название Роксера® Международное непатентованное название:Розувастатин. Форма выпуска:таблетки, покрытые пленочной оболочкой. Состав:1 таблетка, покрытая пленочной оболочкой содержит: Ядро: Активное вещество: 5 мг 10 мг 15 мг Розувастатин кальция 5,21мг 10,42 мг 15,62 мг (что соответствует розувастатину 5 мг 10 мг 15 мг) Вспомогательные вещества: ТАБЛИЦЪ АТХ: Регистрация: Лекарственное средство ЛП-001450 Фармгруппа: гиполипидемическое средство - ГМГ-КоА редуктазы ингибитор. Дата регистрации: 24.01.2012/25.04.2014. Окончание регстрации: 2017-01-24 2017-01-24 . Описание:Таблетки 5 мг:круглые, двояковыпуклые таблетки, покрытые пленочной оболочкой белого цвета, с фаской, на одной стороне маркировка "5", нанесенная методом тиснения. На поперечном разрезе видны два слоя, ядро - белого цвета. Таблетки 10мг:круглые, двояковыпуклые таблетки, покрытые пленочной оболочкой белого цвета, с фаской, на одной стороне маркировка "10", нанесенная методом тиснения. На поперечном разрезе видны два слоя, ядро - белого цвета. Таблетки 15 мг:круглые, двояковыпуклые таблетки, покрытые пленочной оболочкой белого цвета, с фаской, на одной стороне маркировка "15", нанесенная методом тиснения. На поперечном разрезе видны два слоя, ядро - белого цвета. Таблетки 20 мг:круглые, двояковыпуклые таблетки, покрытые пленочной оболочкой белого цвета, с фаской. На поперечном разрезе видны два слоя, ядро - белого цвета. Таблетки 30 мг:капсуловидные двояковыпуклые таблетки, покрытые пленочной оболочкой белого цвета, с риской на обеих сторонах. На поперечном разрезе видны два слоя, ядро - белого цвета. Таблетки 40 мг:капсуловидные, двояковыпуклые таблетки, покрытые пленочной оболочкой белого цвета. На поперечном разрезе видны два слоя, ядро - белого цвета. Упаковка:Таблетки, покрытые пленочной оболочкой, 5 мг, 10 мг, 15 мг, 20 мг, 30 мг и 40 мг. Таблетки 5 мг, 10 мг, 15 мг и 20 мг: по 10 или 14 таблеток в блистер из комбинированного материала ОПА/А1/ПВХ- алюминиевой фольги.1, 2, 3, 6, 9 блистеров (блистер по 10 таблеток) или 1, 2, 4, 6 блистеров (блистер по 14 таблеток) вместе с инструкцией по применению помещают в пачку картонную. Таблетки 30 мг и 40 мг: по 10 или 7 таблеток в блистер из комбинированного материала ОПА/А1/ПВХ- алюминиевой фольги.1, 2, 3, 6, 9 блистеров (блистер по 10 таблеток) или 2, 4, 8, 12 блистеров (блистер по 7 таблеток) вместе с инструкцией по применению помещают в пачку картонную. Срок годности:3 года. Не использовать препарат после истечения срока годности. Владелец рег.удостоверения:КРКА, д.д, Ново место, АО Производитель:KRKA, d.d. Представительство:КРКА, д.д, Ново место, АО
1879 Руб.Роксера таб. п/о 5мг №90
Показания Первичная гиперхолестеринемия по классификации Фредриксона (тип IIа, включая семейную гетерозиготную гиперхолестеринемню) или смешанная гиперхолестеринемия (тип IIb) - в качестве дополнения к диете, когда диета и другие немедикаментозные методы лечения (например, физическиеупражнения, снижение массы тела) оказываются недостаточными- Семейная гомозиготная гиперхолестеринемия - в качестве дополнения к диете и другой липидснижающей терапии (например, ЛПНП-аферез) или в случаях, когда подобная терапия недостаточно эффективна- Гипертриглицеридемия (тип IV по классификации Фредриксона) - в качестве дополнения к диете- Для замедления прогрессирования атеросклероза - в качестве дополнения к диете у пациентов, которым показана терапия для снижения плазменной концентрации общего ХС и ХС-ЛПНП- Первичная профилактика основных сердечно-сосудистых осложнений (инсульта, инфаркта миокарда, артериальной реваскуляризации) у взрослых пациентов без клинических признаков ишемической болезни сердца (ИБС), но с повышенным риском ее развития (возраст старше 50 лет для мужчин и старше 60 лет для женщин, повышенная плазменная концентрация С-реактивного белка (>: 2 мг/л) при наличии, как минимум одного из дополнительных факторов риска, таких как: артериальная гипертензия, низкая плазменная концентрация ХС-ЛПВП, курение, семейный анамнез раннего начала ИБС). Противопоказания Противопоказания При суточной дозе до 30 мг- Повышенная чувствительность к розувастатину или любому из компонентов препарата- заболевания печени в активной фазе (включая стойкое повышение активности "печеночных" трансаминаз и повышение активности "печеночных" трансаминаз в сыворотке крови более чем в 3 раза по сравнению с верхней границей нормы)- тяжелая почечная недостаточность (КК менее 30 мл/мин)- миопатия- одновременный прием циклоспорина- пациенты, предрасположенные к развитию миотоксических осложнений- беременность, период грудного вскармливания- применение у женщин детородного возраста, не пользующихся адекватными методами контрацепции- непереносимость лактозы, дефицит лактазы, синдром глюкозо-галактозной мальабсорбции- возраст до 18 лет. При суточной дозе 30 мг и более- Повышенная чувствительность к розувастатину или любому из компонентов препарата- заболевания печени в активной фазе (включая стойкое повышение активности "печеночных" трансаминаз и повышение активности "печеночных" трансаминаз в сыворотке крови более чем в 3 раза по сравнению с верхней границей нормы)- почечная недостаточность средней и тяжелой степени (КК менее 60 мл/мин)- миопатия- одновременное применение циклоспорина- пациенты, предрасположенные к развитию миотоксических осложнений- беременность, период грудного вскармливания- применение у женщин детородного возраста не пользующихся адекватными методами контрацепции- гипотиреоз- заболевания мышц в анамнезе (в том числе в семейном)- миотоксичность при применении других ингибиторов ГМГ-КоА-редуктазы или фибратов в анамнезе- чрезмерное употребление алкоголя- состояния, которые могут приводить к повышению концентрации розувастатина в плазме крови- одновременное применение фибратов- непереносимость лактозы, дефицит лактазы, синдром глюкозо-галактозной мальабсорбции- пациенты монголоидной расы- возраст до 18 лет. С осторожностью: При суточной дозе до 30 мг. Наличие риска развития миопатии/рабдомиолиза - почечная недостаточность, гипотиреоз, наследственные заболевания мышц в анамнезе (в том числе в семейном) и предшествующий анамнез мышечной токсичности при применении других ингибиторов ГМГ-КоА-редуктазы или фибратов: чрезмерное употребление алкоголя: возраст старше 65 лет: состояния, при которых отмечено повышение плазменной концентрации розувастатина: расовая принадлежность (монголоидная раса - японцы и китайцы): одновременное применение с фибратами: заболевания печени в анамнезе: сепсис: артериальная гипотензия: обширные хирургические вмешательства, травмы, тяжелые метаболические, эндокринные или электролитные нарушения или неконтролируемые судороги, одновременное применение с эзетимибом. При суточной дозе 30 мг и более. Почечная недостаточность легкой степени тяжести (КК более 60 мл/мин): возраст старше 65 лет: заболевания печени в анамнезе: сепсис: артериальная гипотензия: обширные хирургические вмешательства, травмы, тяжелые метаболические, эндокринные или электролитные нарушения или неконтролируемые судороги, одновременное применение с эзетимибом. Беременность Препарат Роксера® противопоказан при беременности и в период лактации. Женщины репродуктивного возраста должны применять адекватные методыконтрацепции. Поскольку холестерин и вещества, синтезируемые из холестерина важны для развития плода, потенциальный риск ингибирования ГМГ-КоА-редуктазы для плода превышает пользу от применения препарата при беременности.В случае наступления беременности в процессе терапии применение препарата должно быть немедленно прекращено. Данные в отношении выделения розувастатина с грудным молоком отсутствуют (известно, что другие ингибиторы ГМГ-КоА-редуктазы способные выделяться с грудным молоком), поэтому в период кормления грудью применение препарата необходимо прекратить. Применение и дозы Внутрь, таблетку не разжевывать и не измельчать, проглатывать целиком, запивая водой, возможен прием в любое время суток независимо от времени приема пищи. До начала терапии препаратом Роксера® пациент должен начать соблюдать стандартную гипохолестеринемическую диету и продолжать соблюдать ее во время лечения. Доза препарата должна подбираться индивидуально в зависимости от целей терапии и терапевтического ответа на лечение, принимая во внимание национальные рекомендации по целевым концентрациям липидов в плазме крови. Рекомендуемая начальная доза для пациентов, начинающих принимать препарат, или для пациентов, переведенных с приема других ингибиторов ГМГ-КоА-редуктазы, должна составлять 5 или 10 мг препарата Роксера® 1 раз в сутки. При одновременном применении препарата с гемфиброзилом, фибратами, никотиновой кислотой в липидснижающих дозах (более 1 г/сут) пациентам рекомендуется начальная доза препарата 5 мг/сут. При выборе начальной дозы следует руководствоваться индивидуальной концентрацией холестерина в плазме крови и принимать во внимание возможный риск развития сердечно-сосудистых осложнений, необходимо также учитывать потенциальный риск развития побочных эффектов. В случае необходимости, доза может быть увеличена через 4 недели. В связи с возможным развитием побочных эффектов при применении дозы 40 мг/сут, по сравнению с более низкими дозами препарата, повышение дозы до максимальной 40 мг/сут следует рассматривать только у пациентов с тяжелой степенью гиперхолестеринемии и с высоким риском развития сердечно-сосудистых осложнений (особенно у пациентов с семейной гиперхолестеринемией), у которых не был достигнут желаемый результат терапии при применении дозы 20 мг/сут, и которые будут находиться под наблюдением врача. Рекомендуется особенно тщательное наблюдение за пациентами, получающими препарат в дозе 40 мг/сут. Не рекомендуется применение дозы 40 мг/сут у пациентов, ранее не обращавшихся к врачу. После 2-4 недель терапии и/или при повышении дозы препарата Роксера® необходим контроль показателей липидного обмена (при необходимости требуется коррекция дозы). Пациенты с почечной недостаточностью У пациентов с почечной недостаточностью легкой или умеренной степени тяжести коррекции дозы не требуется. У пациентов с тяжелой почечной недостаточностью (КК менее 30 мл/мин) применение препарата Роксера® противопоказано. Применение препарата Роксера® в дозе более 30 мг/сут пациентам с почечной недостаточностью умеренной и тяжелой степени (КК менее 60 мл/мин) противопоказано. Пациентам с почечной недостаточностью умеренной степени рекомендуемая начальная доза препарата Роксера® составляет 5 мг/сут. Пациенты с печеночной недостаточностью Препарат Роксера® противопоказан пациентам с заболеваниями печени в активной фазе. Применение у пациентов пожилого возраста Не требуется коррекции дозы. Этнические группыУ пациентов монголоидной расы отмечено увеличение системной экспозиции розувастатина. Для пациентов монголоидной расы рекомендуемая начальная доза препарата Роксера® составляет 5 мг/сут, применение препарата Роксера® дозе 40 мг противопоказано. Генетический полиморфизмУ носителей генотипов SLC01B1 (ОАТР1В1) с.521СС и ABCG2 (BCRP) с.421АА отмечалось увеличение экспозиции (AUC) розувастатина по сравнению с носителями генотипов SLCO1B1 с.521ТТ и ABCG2 с.421СС. Для пациентов-носителей генотипов с.521СС или с.421АА рекомендуемая максимальная доза препарата Роксера® составляет 20 мг 1 раз в сутки. Пациенты, предрасположенные к миотоксическим осложнениям Применение препарата Роксера® в дозе 40 мг пациентам, предрасположенным к развитию миотоксических осложнений противопоказано. При необходимости применения доз К) и 20 мг/сут рекомендуемая начальная доза для данной группы пациентов составляет 5 мг/сут. Сопутствующая терапия. Розувастатин связывается с различными транспортными белками (в частности, с ОАТР1В1 и BCRP). При одновременном применении препарата Роксера с лекарственными препаратами (такими как циклоспорин, некоторые ингибиторы протеазы вируса иммунодефицита человека (ВИЧ), включая комбинацию ритонавира с атазапавиром, лопипавиром и/или типранавиром), повышающими концентрацию розувастатина в плазме крови за счет взаимодействия с траспортными белками, может повышаться риск развития миопатии (включая рабдомиолиз). Следует ознакомиться с инструкцией по применению вышеуказанных препаратов перед их назначением одновременно с препаратом Роксера®. В таких случаях следует оценить возможность применения альтернативной терапии или временного прекращения приема препарата Роксера®. При необходимости применения указанных выше препаратов, следует оценить соотношение пользы и риска сопутствующей терапии препаратом Роксера® и рассмотреть возможность снижения его дозы. Побочные эффекты и передозировка Побочные эффекты: Побочные эффекты, наблюдаемые при приеме препарата Роксера®, обычно выражены незначительно и проходят самостоятельно. Как и при применении других ингибиторов ГМГ-КоА-редуктазы, частота возникновения Абсорбция и распределение Максимальная концентрация розувастатина в плазме крови достигается приблизительно через 5 ч после приема внутрь. Абсолютная биодоступность составляет примерно 20 %. Метаболизируется преимущественно печенью, которая является основным органом, синтезирующим холестерин и метаболизирующим ХС-ЛПНП. Объем распределения розувастатина составляет примерно 134 л. Приблизительно 90 % розувастатина связывается с белками плазмы крови, в основном с альбумином. Метаболизм Подвергается ограниченному метаболизму (около 10 %). Розувастатин является побочных эффектов носит, в основном, дозозависимый характер. Классификация частоты развития побочных эффектов Всемирной организации здравоохранения (ВОЗ): очень часто >: 1/10 часто от >: 1/100 до <: 1/10 нечасто от >: 1/1000 до <: 1/100 редко от >: 1/10000 до <: 1/1000 очень редко <: 1/10000 частота неизвестна не может быть оценена на основе имеющихся данных. Нарушения со стороны крови и лимфатической системы: частота неизвестна: тромбоцитопения. Нарушения со стороны иммунной системы: редко: реакции повышенной чувствительности, включая ангионеврогический отёк. Нарушения со стороны эндокринной системы: часто: сахарный диабет 2-го типа. Нарушения со стороны нервной системы: часто: головная боль, головокружение: очень редко: потеря или снижение памяти: частота неизвестна: периферическая нейропатия. Нарушения со стороны дыхательной системы, органов грудной клетки и средостения:частота неизвестна: кашель, одышка. Нарушения со стороны пищеварительной системы:часто: запор, тошнота, боль в животе:редко: панкреатит:очень редко: желтуха, гепатит:частота неизвестна: диарея. При применении розувастатина наблюдается дозозависимое повышение активности "печеночных" трансамнназ в плазме крови у незначительного числа пациентов. В большинстве случае оно незначительно, бессимптомно и временно. Нарушения со стороны кожи и подкожных тканей:нечасто: кожный зуд, кожная сыпь, крапивница:частота неизвестна: синдром Стивенса-Джонсона. Нарушения со стороны скелетно- мышечной и соединительной ткани:часто: миалгия:редко: миопатия (включая миозит), рабдомиолиз (с острой почечной недостаточностью или без нее):очень редко: артралгия:частота неизвестна: иммуноопосредованная некротизирующая миопатия. Дозозависимое повышение активности креатинфосфокиназы (КФК) в плазме крови наблюдается у небольшого числа пациентов, принимавших розувастатин. В большинстве случаев оно является незначительным, бессимптомным и временным. В случае повышения активности КФК в плазме крови более чем в 5 раз выше верхней границы нормы терапию следует приостановить. Нарушения со стороны почек и мочевыводящих путей:У пациентов, получающих терапию розувастатином, может выявляться протеинурия. Изменение количества белка в моче (от отсутствия или следовых количеств до + или больше) наблюдается менее чем у 1 % пациентов, получающих 10-20 мг препарата, и у приблизительно 3 % пациентов, получающих 40 мг препарата. Незначительное изменение количества белка в моче отмечалось при приеме дозы 20 мг. В большинстве случаев протеинурия уменьшается или исчезает в процессе терапии и не означает возникновенияострого или прогрессирования существующего заболевания почек. Очень редко: гематурия. Нарушения со стороны половых органов и молочной железы:частота неизвестна: гинекомастия. Общие расстройства и нарушения в месте введения: часто: астенический синдром: частота неизвестна: периферические отеки. Лабораторные показатели:При применении розувастатина также наблюдались следующие изменения лабораторных показателей: гипергликемия, повышение концентрации билирубина в плазме крови, активности гамма-глутамилтраиспептидазы, щелочной фосфатазы в плазме крови, изменение сывороточной концентрации гормонов щитовидной железы. При применении некоторых ингибиторов ГМГ-КоА-редуктазы (статинов) сообщалось о следующих побочных эффектах: депрессия, нарушения сна, включая бессонницу и "кошмарные" сновидения, сексуальная дисфункция, повышение концентрации гликозилированного гемоглобина. Сообщалось о единичных случаях интерстициального заболевания легких, особенно при длительном применении препаратов (см. раздел "Особые указания"). Передозировка: Клиническая картина передозировки не описана. При единовременном приеме нескольких суточных доз препарата фармакокинетические параметры розувастатина не изменяются. Лечение передозировки - симптоматическое, необходим контроль функции печени и активности КФК: специфического антидота не существует, гемодиализ неэффективен. Взаимодействие с другими ЛС: Влияние применения других препаратов на розувастатин Ингибиторы транспортных белков Розувастатин является субстратом для некоторых транспортных белков, в частности, ОАТР1В1 и BCRP. Одновременное применение препаратов, которые являются ингибиторами этих транспортных белков,может сопровождаться увеличением концентрации розувастатина в плазме крови и повышенным риском развития миопатии (см. таблицу 1, разделы "Способ применения и дозы", "Особые указания"). Циклоспорин При одновременном применении розувастатина и циклоспорина, AUC розувастатина в среднем в 7 раз выше значения, которое отмечается у здоровых добровольцев (см. таблицу 1). Одновременноеприменение с розувастатином не влияет на концентрацию циклоспорина в плазме крови. Применение розувастатина противопоказано пациентам, принимающим циклоспорин (см. раздел "Противопоказания"). Ингибиторы протеазы ВИЧ Одновременное применение ингибиторов протеазы ВИЧ может значительно повышать экспозицию розувастатина (см. таблицу 1). Одновременное применение 20 мг розувастатина и комбинации двух ингибиторов протеазы ВИЧ (400 мг лопинавира/100 мгритонавира) сопровождается повышением равновесных AUC(0-24) и Сmах розувастатина в 2 и 5 раз, соответственно. Поэтому одновременный прием розувастатина и ингибиторов протеазы ВИЧ не рекомендуется (см. раздел "Способ применения и дозы", таблицу 1). Гемфиброзил и другие гиполипидемические средства Одновременное применение розувастатина и гемфиброзила приводит к увеличению Сmax и AUC розувастатина в плазме крови в 2 раза (см. раздел "Особые указания"). Основываясь на данных по специфическому взаимодействию, не ожидается фармакокинетически значимого взаимодействия с фенофибратом, возможно фармакодинамическое взаимодействие. Гемфиброзил, фенофибрат, другие фибраты и липидснижающие дозы никотиновой кислоты (дозы большие или эквивалентные 1 г/сут) увеличивали риск возникновения миопатии при одновременном применении с ингибиторами ГМГ-КоА-редуктазы, возможно в связи с тем, что они могут вызывать миопатию при применении в монотерапии (см. раздел "Особые указания"). Одновременное применение фибратов и розувастатина в суточной дозе 30 мг противопоказано. У таких пациентов терапия должна начинаться с дозы 5 мг/сут (см. разделы "Противопоказания", "Способ применения и дозы", "Особые указания"). Эзетимиб Одновременное применение розувастатина в дозе 10 мг и эзетимиба в дозе 10 мг сопровождалось увеличением AUC розувастатина у пациентов с гиперхолестеринемией (см. таблицу 1). Нельзя исключить фармакодинамическое взаимодействие между розувастатином и эзегимибом, проявляющееся увеличением риска развития нежелательных реакций. Антациды Одновременное применение розувастатина и антацидов, содержащих алюминия и магния гидроксид, приводит к снижению плазменной концентрации розувастатина примерно на 50 %. Данный эффект выражен слабее, если антациды применяются через 2 ч после приема розувастатина. Клиническое значение подобного взаимодействия не изучалось. Эритромицин Одновременное применение розувастатина и эритромицина приводит к уменьшению AUC(o-t) розувастатина на 20 % и его Сmах на 30 %. Подобное взаимодействие может возникать в результате усиления моторики кишечника, вызываемого применением эритромицина. Изоферменты системы цитохрома Р450 Результаты исследований in vivo и in vitro показали, что розувастатин не является ни ингибитором, ни индуктором изоферментов системы цитохрома Р450. Кроме того, розувастатин является слабым субстратом для этой системы изоферментов. Поэтому неожидается взаимодействия розувастатина с другими лекарственными средствами (ЛС) на уровне метаболизма с участием изоферментов системы цитохрома Р450. Клинически значимого взаимодействия между розувастатином и флуконазолом (ингибитором изоферментов CYP2C9 и CYP3A4) и кетоконазолом (ингибитором изоферментов CYP2A6 и CYP3A4) не отмечено. Фузидовая кислота Исследований по изучению взаимодействия розувастатина и фузидовой кислоты не проводилось. Как и при приеме других статинов, были полученыпостмаркетинговые сообщения о случаях рабдомиолиза при одновременномприменении розувастатина и фузидовой кислоты. Необходимо пристально наблюдать за пациентами. При необходимости, возможно временное прекращение приема розувастатина. Взаимодействие с лекарственными средствами, которое требует коррекции дозы розувастатина (см. таблицу 1) Дозу препарата Роксера® следует корректировать при необходимости его одновременного применения с ЛC, увеличивающими экспозицию розувастатина. Если ожидается увеличение экспозиции в 2 раза и более, начальная доза препарата Роксера® должна составлять 5 мг 1 раз в сутки. Также следует корректировать максимальную суточную дозу препарата Роксера®, чтобы ожидаемая экспозиция розувастатина не превышала таковую для дозы 40 мг, принимаемой без одновременного назначения ЛC, взаимодействующих с розувастатином. Например, максимальная суточная доза препарата Роксера® при одновременном применении с гемфиброзилом составляет 20 мг (увеличение экспозиции в 1,9 раза), с ритонавиром/атазанавиром - 10 мг (увеличение экспозиции в 3,1 раза). Таблица 1. Влияние сопутствующей терапии на экспозицию розувастатина (AUC, данные приведены в порядке убывания) - результаты опубликованных клинических исследований ТАБЛИЦЪ Влияние применения розувастатина на другие препараты Антагонисты витамина К Как и в случае других ингибиторов ГМГ- КоА-редукгазы, начало терапии розувастатином или увеличение его дозы у пациентов, принимающих одновременно антагонисты витамина К (например, варфарин), может приводить к увеличению Международного нормализованного отношения (MHO). Отмена розувастатина или снижение его дозы может приводить к уменьшению MHO. В таких случаях рекомендуется мониторинг MHO. Контрацептивы для приема внутрь/заместительная гормональная терапия (ЗГТ) Одновременное применение розувастатина и контрацептивов для приема внутрь увеличивает AUC этинилэстрадиола и норгестрела на 26 % и 34 %, соответственно. Такое увеличение концентрации в плазме крови должно учитываться при подборе дозы гормональных контрацептивов. Фармакокинетические данные по одновременному применению розувастатина и ЗГТ отсутствуют, следовательно, нельзя исключить аналогичного эффекта и при применении данной комбинации. Однако подобная комбинация широко применялась во время проведения клинических исследований и хорошо переносилась пациентами. Другие лекарственные средства Клинически значимого взаимодействия розувастатина с дигоксином не ожидается. Фармакологическое действие и фармакокинетика Препарат Роксера® - гиполипидемическое средство. Действующее вещество препарата -розувастатин, является селективным, конкурентным ингибитором ГМГ-КоА редуктазы, фермента, превращающего З-гидрокси-З-метилглутарилкофермент А в мевалоновую кислоту - предшественник холестерина. Основной мишенью действия розувастатина является печень, где происходит синтез холестерина (ХС) и катаболизм липопротеинов низкой плотности (ЛПНП). Увеличивает число печеночных рецепторов к ЛПНП на поверхности клеток, повышая захват и катаболизм ЛПНП, что в свою очередь приводит к ингибированию синтеза липопротеинов очень низкой плотности (ЛПОНП), уменьшая тем самым общее количество ЛПНП и ЛПОНП. Розувастатин снижает повышенные плазменные концентрации холестерина ЛПНП (ХС-ЛПНП), общего холестерина, триглицеридов (ТГ), повышает концентрацию холестерина липопротеинов высокой плотности (ХС-ЛПВП). Он также снижает концентрацию аполипопротеина В (АпоВ), ХС-неЛПВП, ХС-ЛПОНП, ТГ-ЛПОНП и увеличивает концентрацию аполипопротеина A-I в плазме крови. Розувастатин снижает соотношение ХС-ЛПНП/ХС-ЛПВП, общий ХС/ХС-ЛПВП и ХС-неЛПВП/ ХС-ЛПВП и соотношение АпоВ/АпоА-1. Терапевтический эффект развивается в течение одной недели после начала терапии, через 2 недели лечения достигает 90 % от максимально возможного эффекта. Максимальный терапевтический эффект обычно достигается к 4 неделе терапии и поддерживается при дальнейшем регулярном приёме препарата. Клиническая эффективность. Розувастатин эффективен у взрослых пациентов с гиперхолестеринемией с сопутствующей гипертриглицеридемией или без нее, вне зависимости от расовой принадлежности, пола или возраста, в том числе у пациентов с сахарным диабетом или семейной гиперхолестеринемией.У 80 % пациентов с гиперхолестеринемией Па и lib типа по Фредриксону (средняя исходная концентрация ХС-ЛПНП примерно 4,8 ммоль/л) на фоне приема розувастатина в дозе 10 мг концентрация ХС-ЛПНП достигает значений менее ммоль/л. У пациентов с гомозиготной семейной гиперхолестеринемией розувастатин применялся в дозах 20-40 мг, среднее снижение составило 22 %. Аддитивный эффект отмечается в комбинации с фенофибратом в отношении содержания ТГ и с никотиновой кислотой в липидснижающих дозах в отношении концентрации ХС-ЛПВП. Фармакокинетика: Абсорбция и распределение Максимальная концентрация розувастатина в плазме крови достигается приблизительно через 5 ч после приема внутрь. Абсолютная биодоступность составляет примерно 20 %. Метаболизируется преимущественно печенью, которая является основным органом, синтезирующим холестерин и метаболизирующим ХС-ЛПНП. Объем распределения розувастатина составляет примерно 134 л. Приблизительно 90 % розувастатина связывается с белками плазмы крови, в основном с альбумином. Метаболизм Подвергается ограниченному метаболизму (около 10 %). Розувастатин является неспецифическим субстратом цитохрома Р450. Основным изоферментом, участвующим в метаболизме розувастатина, является изоферменг CYP2C9. Изоферменты CYP2C19, CYP3A4, CYP2D6 вовлечены в метаболизм в меньшей степени. Основными выявленными метаболитами являются N-десметилрозувастатин и лактоновые метаболиты. N-десметилрозувастатин примерно на 50 % менее активен, чем розувастатин, лактоновые метаболиты фармакологически неактивны. Более 90 % фармакологической активности по ингибированию плазменной ГМГ-КоА-редуктазы обеспечивается розувастатином, остальное - его метаболитами. Выведение Около 90 % дозы розувастатина выводится в неизмененном виде через кишечник (включая абсорбированный и неабсорбированный розувастатин). Оставшаяся часть выводится почками. Период полувыведения (Т1/2) из плазмы крови составляет примерно 19 ч (не изменяется при увеличении дозы препарата). Средний геометрический плазменный клиренс - 50 л/ч (коэффициент вариации - 21,7 %). Как и в случае других ингибиторов ГМГ-КоА-редуктазы, в процесс "печеночного" захвата розувастатина вовлечен мембранный переносчик холестерина, выполняющий важную роль в "печеночной" элиминации розувастатина. Линейность Системная экспозиция розувастатина увеличивается пропорционально дозе. Фармакокинетические параметры не изменяются при ежедневном приеме. Особые группы пациентов Возраст и пол Пол и возраст не оказывают клинически значимого влияния на фармакокинетику розувастатина. Этнические группы. Фармакокинетические исследования показали приблизительно двукратное увеличение медианы AUC (площади под кривой "концентрация-время") и максимальной концентрации (Сmах) в плазме крови розувастатина у пациентов монголоидной расы (японцев, китайцев, филиппинцев, вьетнамцев и корейцев) по сравнению с пациентами европеоидной расы: у индийцев показано увеличение медианы AUC и Сmах в 1,3 раза. Фармакокинетический анализ не выявил клинически значимых различий в фармакокинетике среди пациентов европеоидной и негроидной рас. Почечная недостаточностьУ пациентов с почечной недостаточностью легкой или умеренной степени тяжести величина плазменной концентрации розувастатина или N-десметилрозувастатина существенно не меняется. У пациентов с тяжелой почечной недостаточностью (клиренс креатинина (КК) менее 30 мл/мин) концентрация розувастатина в плазме крови в 3 раза выше, а концентрация N-десмегилрозувастатина в 9 раз выше, чем у здоровых добровольцев. Концентрация розувастатина в плазме крови у пациентов, находящихся на гемодиализе, примерно на 50 % выше, чем у здоровых добровольцев. Печеночная недостаточность У пациентов с печеночной недостаточностью 7 баллов и ниже по шкале Чайлд-Пью не выявлено увеличения системной экспозиции розувастатина. У двух пациентов с печеночной недостаточностью 8-9 баллов по шкале Чайлд-Пью отмечено увеличение системной экспозиции, по крайней мере, в 2 раза. Опыт применения розувастатина у пациентов с печеночной недостаточностью выше 9 баллов по шкале Чайлд-Пью отсутствует. Генетический полиморфизм Ингибиторы ГМГ-КоА-редуктазы, в том числе розувастатин, связываются с транспортными белками ОАТР1В1 (полипептид транспорта органических анионов, участвующий в захвате статинов гепатоцитами) и BCRP (эффлюксный транспортер). У носителей генотипов SLCO1B1 (ОАТР1В1) с.521СС и ABCG2 (BCRP) c.421АА отмечалось увеличение экспозиции (AUC) розувастатина в 1,6 и 2,4 раза, соответственно, по сравнению с носителями генотипов SLCO1B1 c.521ТТ и ABCG2 с.421СС. Особые указания Нарушение функции почек У пациентов, получавших высокие дозы розувастатина (в частности 40 мг/сут), наблюдалась канальцевая протеинурия, которая выявлялась при помощи тест- полосок и в большинстве случаев была периодической или кратковременной. Такая протеинурия не свидетельствует об остром или прогрессировании сопутствующего заболевания почек. Частота серьезных нарушений функции почек, отмеченная при постмаркетинговом изучении розувастатина, выше при приёме дозы 40 мг/сут. У пациентов, принимающих препарат Роксера® в дозе 30 или 40 мг/сут, рекомендуется контролировать показатели функции почек во время лечения (не реже чем 1 раз в 3 месяца). Влияние на опорно-двигательный аппарат При применении розувастатина во всех дозах, но в особенности в дозах, превышающих 20 мг/сут, сообщалось о следующих воздействиях на опорно- двигательный аппарат: миалгия, миопатия, в редких случаях рабдомиолиз. Отмечены очень редкие случаи рабдомиолиза при одновременном применении ингибиторов ГМГ-КоА-редуктазы и эзетимиба. Такая комбинация должна применяться с осторожностью, так как нельзя исключить фармакодинамического взаимодействия. Как и в случае других ингибиторов ГМГ-КоА-редуктазы, частота рабдомиолиза при постмаркетинговом применении препарата Роксера® выше при применении дозы 40 мг/сут. Определение активности КФК Активность КФК нельзя определять после интенсивных физических нагрузок и при наличии других возможных причин повышения ее активности: это может привести к неверной интерпретации полученных результатов. В случае если исходная активность КФК существенно превышена (в 5 раз выше верхней границы нормы), через 5-7 дней следует провести повторный анализ. Нельзя начинать терапию, если результаты повторного анализа подтверждают исходную высокую активность КФК (более чем 5-кратное превышение верхней границы нормы). Перед началом терапии В зависимости от суточной дозы препарат Роксера® должен назначаться с осторожностью пациентам с имеющимися факторами риска миопатии/рабдомиолиза или применение препарата противопоказано (см. разделы "Противопоказания" и "С осторожностью"). К таким факторам относятся: - нарушение функции почек: - гипотиреоз: - заболевания мышц в анамнезе (в том числе в семейном): - миотоксические явления при приёме других ингибиторов ГМГ-КоА-редуктазы или фибратов в анамнезе: - чрезмерное употребление алкоголя: - возраст старше 65 лет: - состояния, при которых может повышаться концентрация розувастатина в плазме крови- одновременное применение фибратов. У таких пациентов необходимо оценить риск и возможную пользу терапии. Также рекомендуется проводить клинический мониторинг. Если исходная активность КФК выше более чем в 5 раз по сравнению с верхней границей нормы, терапию препаратом Роксера® начинать нельзя. В период терапии препаратом Следует проинформировать пациента о необходимости немедленного сообщения врачу в случае неожиданного появления мышечных болей, мышечной слабости или спазмов, особенно в сочетании с недомоганием и лихорадкой. У таких пациентов следует определять активность КФК. Терапия должна быть прекращена, если активность КФК значительно увеличена (более чем в 5 раз по сравнению с верхней границей нормы) или если симптомы со стороны мышц резко выражены и вызывают ежедневный дискомфорт (даже, если активность КФК не более чем в 5 раз превышает верхнюю границу нормы). Если симптомы исчезают и активность КФК возвращается к норме, следует рассмотреть вопрос о возобновлении применения препарата Роксера® или других ингибиторов ГМГ-КоА-редуктазы в меньших дозах при тщательном медицинском наблюдении. Контроль активности КФК при отсутствии симптомов нецелесообразен. Отмечены очень редкие случаи иммуноопосредованной некротизирующей миопатии с клиническими проявлениями в виде стойкой слабости проксимальных мышц и повышения активности КФК в сыворотке крови во время терапии или при прекращении применения ингибиторов ГМГ-КоА-редуктазы, в том числе розувастатина. Может потребоваться проведение дополнительных исследований мышечной и нервной системы, серологических исследований, а также терапия иммунодепрессивными средствами. Признаков увеличения воздействия на скелетную мускулатуру при приеме розувастатина и сопутствующей терапии не отмечено. Однако сообщалось об увеличении числа случаев миозита и миопатии у пациентов, принимавших другие ингибиторы ГМГ-КоА-редуктазы в сочетании с производными фиброевой кислоты (например, гемфиброзил), циклоспорином, никотиновой кислотой в липидснижающих дозах (более 1 г/сутки), противогрибковыми средствами производными азола, ингибиторами протеазы ВИЧ и макролидными антибиотиками. При одновременном применении с некоторыми ингибиторами ГМГ-КоА- редуктазы гемфиброзил увеличивает риск развития миопатии. Таким образом, одновременное применение препарата Роксера® и гемфиброзила не рекомендуется. Преимущества дальнейшего изменения плазменной концентрации липидов при комбинированном применении препарата Роксера® с фибратами или никотиновой кислотой в липидснижающих дозах должны быть тщательно взвешены с учётом возможного риска. Препарат Роксера® в дозе 30 мг/сут противопоказан для комбинированной терапии с фибратами. В связи с увеличением риска рабдомиолиза препарат Роксера® не следует применять пациентам с острыми состояниями, которые могут привести к миопатии или состояниям, предрасполагающим к развитию почечной недостаточности (например, сепсис, артериальная гипотензия, обширные хирургические вмешательства, травмы, тяжелые метаболические, эндокринные и электролитные нарушения или неконтролируемые судороги). Печень В зависимости от суточной дозы препарат Роксера® должен с осторожностью применяться у пациентов с чрезмерным употреблением алкоголя и/или имеющих в анамнезе заболевания печени или его применение противопоказано (см. разделы "Противопоказания" и "С осторожностью"). Рекомендуется проводить определение функциональных проб печени до начала терапии и через 3 месяца после ее начала. Применение препарата Роксера® следует прекратить или уменьшить дозу препарата, если активность "печеночных" трансаминаз в сыворотке крови в 3 раза превышает верхнюю границу нормы.У пациентов с гиперхолестеринемией вследствие гипотиреоза или нефротического синдрома до начала лечения препаратом Роксера® должна проводиться терапия основных заболеваний. Этнические особенностиВ ходе фармакокинетических исследований у представителей монголоидной расы по сравнению с представителями европеоидной расы отмечено увеличение плазменной концентрации розувастатина. Препарат Роксера® содержит лактозу, в связи с чем его не следует применять пациентам с непереносимостью лактозы, дефицитом лактазы, синдромом глюкозо- галактозной мальабсорбции. Интерстициалъное заболевание легких. При применении некоторых ингибиторов ГМГ-КоА-редуктазы, особенно в течение длительного времени, сообщалось о единичных случаях интерстициального заболевания легких. Проявлениямизаболевания могут являться одышка, непродуктивный кашель и ухудшение общего самочувствия (слабость, снижение веса и лихорадка). При подозрении на интерстициальное заболевание легких следует прекратить терапию ингибиторами ГМГ-КоА- редуктазы. Сахарный диабет 2-го типаУ пациентов с концентрацией глюкозы от 5,6 до 6,9 ммоль/л терапия розувастатином ассоциировалась с повышенным риском развития сахарного диабета 2-го типа. Влияние на способность управлять транспортными средствами: исследования по изучению влияния препарата Роксера® на способность управлять транспортными средствами и работу с механизмами не проводились. Тем не менее, учитывая возможность развития головокружения и др. побочных эффектов, необходимо соблюдать осторожность при управлении транспортными средствами и другими механизмами, требующими повышенной концентрации внимания и быстроты психомоторных реакций. Условия хранения и отпуска из аптек Условия хранения:При температуре не выше 25 °С, в оригинальной упаковке. Хранить в недоступном для детей месте. Отпуск из аптек: По рецепту Регистрационные данные Торговое название Роксера® Международное непатентованное название:Розувастатин. Форма выпуска:таблетки, покрытые пленочной оболочкой. Состав:1 таблетка, покрытая пленочной оболочкой содержит: Ядро: Активное вещество: 5 мг 10 мг 15 мг Розувастатин кальция 5,21мг 10,42 мг 15,62 мг (что соответствует розувастатину 5 мг 10 мг 15 мг) Вспомогательные вещества: ТАБЛИЦЪ АТХ: Регистрация: Лекарственное средство ЛП-001450 Фармгруппа: гиполипидемическое средство - ГМГ-КоА редуктазы ингибитор. Дата регистрации: 24.01.2012/25.04.2014. Окончание регстрации: 2017-01-24 2017-01-24 . Описание:Таблетки 5 мг:круглые, двояковыпуклые таблетки, покрытые пленочной оболочкой белого цвета, с фаской, на одной стороне маркировка "5", нанесенная методом тиснения. На поперечном разрезе видны два слоя, ядро - белого цвета. Таблетки 10мг:круглые, двояковыпуклые таблетки, покрытые пленочной оболочкой белого цвета, с фаской, на одной стороне маркировка "10", нанесенная методом тиснения. На поперечном разрезе видны два слоя, ядро - белого цвета. Таблетки 15 мг:круглые, двояковыпуклые таблетки, покрытые пленочной оболочкой белого цвета, с фаской, на одной стороне маркировка "15", нанесенная методом тиснения. На поперечном разрезе видны два слоя, ядро - белого цвета. Таблетки 20 мг:круглые, двояковыпуклые таблетки, покрытые пленочной оболочкой белого цвета, с фаской. На поперечном разрезе видны два слоя, ядро - белого цвета. Таблетки 30 мг:капсуловидные двояковыпуклые таблетки, покрытые пленочной оболочкой белого цвета, с риской на обеих сторонах. На поперечном разрезе видны два слоя, ядро - белого цвета. Таблетки 40 мг:капсуловидные, двояковыпуклые таблетки, покрытые пленочной оболочкой белого цвета. На поперечном разрезе видны два слоя, ядро - белого цвета. Упаковка:Таблетки, покрытые пленочной оболочкой, 5 мг, 10 мг, 15 мг, 20 мг, 30 мг и 40 мг. Таблетки 5 мг, 10 мг, 15 мг и 20 мг: по 10 или 14 таблеток в блистер из комбинированного материала ОПА/А1/ПВХ- алюминиевой фольги.1, 2, 3, 6, 9 блистеров (блистер по 10 таблеток) или 1, 2, 4, 6 блистеров (блистер по 14 таблеток) вместе с инструкцией по применению помещают в пачку картонную. Таблетки 30 мг и 40 мг: по 10 или 7 таблеток в блистер из комбинированного материала ОПА/А1/ПВХ- алюминиевой фольги.1, 2, 3, 6, 9 блистеров (блистер по 10 таблеток) или 2, 4, 8, 12 блистеров (блистер по 7 таблеток) вместе с инструкцией по применению помещают в пачку картонную. Срок годности:3 года. Не использовать препарат после истечения срока годности. Владелец рег.удостоверения:КРКА, д.д, Ново место, АО Производитель:KRKA, d.d. Представительство:КРКА, д.д, Ново место, АО
979 Руб.Показания Первичная гиперхолестеринемия по классификации Фредриксона (тип IIа, включая семейную гетерозиготную гиперхолестеринемию) или смешанная гиперхолестеринемия (тип IIb) в качестве дополнения к диете, когда диета и другие немедикаментозные методы лечения (например, физические упражнения, снижение массы тела) оказываются недостаточными- Семейная гомозиготная гиперхолестеринемия в качестве дополнения к диете и другой липидснижающей терапии (например, ЛПНП-аферез), или в случаях, когда подобная терапия недостаточно эффективна- Гипертриглицеридемия (тип IV по классификации Фредриксона) в качестве дополнения к диете- Для замедления прогрессирования атеросклероза в качестве дополнения к диете у пациентов, которым показана терапия для снижения концентрации общего ХС и ХС-ЛПНП- Первичная профилактика основных сердечно-сосудистых осложнений (инсульта, инфаркта, артериальной реваскуляризации) у взрослых пациентов без клинических признаков ИБС, но с повышенным риском ее развития (возраст старше 50 лет для мужчин и старше 60 лет для женщин, повышенная концентрация С-реактивного белка (более 2 мг/л) при наличии, как минимум одного из дополнительных факторов риска, таких как артериальная гипертензия, низкая концентрация ХС-ЛПВП, курение, семейный анамнез раннего начала ИБС). Противопоказания Противопоказания Для препарата Розувастатин-СЗ в суточной дозе 5 мг, 10 мг и 20 мг- повышенная чувствительность к розувастатину или любому из компонентов препарата- непереносимость лактозы, дефицит лактазы или глюкозо-галактозная мальабсорбция (препарат содержит лактозу)- детский возраст до 18 лет- заболевания печени в активной фазе, включая стойкое повышение сывороточной активности трансаминаз и любое повышение активности трансаминаз в сыворотке крови (более чем в 3 раза по сравнению с верхней границей нормы)- тяжелая почечная недостаточность (КК менее 30 мл/мин.)- миопатия- одновременный прием циклоспорина- у женщин: беременность: период грудного вскармливания, отсутствие адекватных методов контрацепции- повышение концентрации креатинфосфокиназы (КФК) в крови более чем в 5 раз по сравнению с верхней границей нормы- совместное применение с ингибиторами ВИЧ-протеаз- пациентам, предрасположенным к развитию миотоксических осложнений. Для препарата Розувастатин-СЗ в суточной дозе 40 мг- повышенная чувствительность к розувастатину или любому из компонентов препарата- непереносимость лактозы, дефицит лактазы или глюкозо-галактозная мальабсорбция (препарат содержит лактозу)- детский возраст до 18 лет- одновременный прием циклоспорина- у женщин: беременность, период грудного вскармливания, отсутствие адекватных методов контрацепции- повышение концентрации креатинфосфокиназы (КФК) в крови более чем в 5 раз по сравнению с верхней границей нормы- совместное применение с ингибиторами ВИЧ-протеаз- почечная недостаточность средней и тяжелой степени (КК менее 60 мл/мин.)- заболевания печени в активной фазе, включая стойкое повышение сывороточной активности трансаминаз и любое повышение активности трансаминаз в сыворотке крови (более чем в 3 раза по сравнению с верхней границей нормы) пациентам с факторами риска развития миопатии/рабдомиолиза, а именно- гипотиреоз- миотоксичность на фоне приема других ингибиторов ГМГ-КоА-редуктазы или фибратов в анамнезе- чрезмерное употребление алкоголя- состояния, которые могут приводить к повышению плазменной концентрации розувастатина- одновременный прием фибратов- миопатия- личный или семейный анамнез мышечных заболеваний - пациентам монголоидной расы С осторожностью: Для препарата Розувастатин-СЗ в суточной дозе 5 мг, 10 мг и 20 мг: Наличие риска развития миопатии/рабдомиолиза - почечная недостаточность, гипотиреоз, личный или семейный анамнез наследственных мышечных заболеваний и предшествующий анамнез мышечной токсичности при использовании других ингибиторов ГМГ-КоА-редуктазы или фибратов: чрезмерное употребление алкоголя: возраст старше 65 лет: состояния, при которых отмечено повышение плазменной концентрации розувастатина: расовая принадлежность (монголоидная раса): одновременное назначение с фибратами (см. раздел "Фармакокинетика"): заболевания печени в анамнезе: сепсис: артериальная гипотензия: обширные хирургические вмешательства, травмы, тяжелые метаболические, эндокринные или электролитные нарушения или неконтролируемые судорожные припадки. Одновременное применение с колхицином и с эзетимибом (см. раздел "Взаимодействие с другими лекарственными препаратами"). Для препарата Розувастатин-СЗ в суточной дозе 40 мг: Почечная недостаточность легкой степени тяжести (КК более 60 мл/мин): возраст старше 65 лет: заболевания печени в анамнезе: сепсис: артериальная гипотензия: обширные хирургические вмешательства, травмы, тяжелые метаболические, эндокринные или электролитные нарушения или неконтролируемые судорожные припадки. Одновременное применение с колхицином и с эзетимибом (см. раздел "Взаимодействие с другими лекарственными препаратами"). Пациенты с печеночной недостаточностью Данные или опыт применения препарата у пациентов с более чем 9-ю баллами по шкале Чайлд-Пью отсутствует (см. разделы "Фармакодинамика" и "Особые указания"). Беременность Розувастатин-СЗ противопоказан при беременности и в период грудного вскармливания. Женщины репродуктивного возраста должны применять адекватные методы контрацепции. Поскольку холестерин и другие продукты биосинтеза холестерина важны для развития плода, потенциальный риск ингибирования ГМГ-КоА-редуктазы превышает пользу от применения препарата у беременных. В случае возникновения беременности в процессе терапии прием препарата должен быть прекращен немедленно. Данные в отношении выделения розувастатина с грудным молоком отсутствуют, поэтому в период грудного вскармливания прием препарата необходимо прекратить (см. раздел "Противопоказания"). Применение и дозы Внутрь, не разжевывать и не измельчать таблетку, проглатывать целиком, запивая водой. Препарат может назначаться в любое время суток независимо от приема нищи. До начала терапии препаратом Розувастатин-СЗ пациент должен начать соблюдать стандартную гипохолестеринемическую диету и продолжать соблюдать ее во время лечения. Доза препарата должна подбираться индивидуально в зависимости от целей терапии и терапевтического ответа на лечение, принимая во внимание текущие рекомендации по целевым концентрациям липидов. Рекомендуемая начальная доза для пациентов, начинающих принимать препарат, или для пациентов, переведенных с приема других ингибиторов ГМГ-КоА-редуктазы, должна составлять 5 мг или 10 мг препарата Розувастатин-СЗ 1 раз в сутки. При выборе начальной дозы следует руководствоваться индивидуальным содержанием холестерина и принимать во внимание возможный риск сердечнососудистых осложнений, а также необходимо оценивать потенциальный риск развития побочных эффектов. В случае необходимости, доза может быть увеличена до большей через 4 недели (см. раздел "Фармакодинамика").В связи с возможным развитием побочных эффектов при приеме дозы 40 мг, по сравнению с более низкими дозами препарата (см. раздел "Побочное действие"), увеличение дозы до 40 мг, после дополнительного приема дозы выше рекомендуемой начальной дозы в течение 4-х недель терапии, может проводиться только у пациентов с тяжелой степенью гиперхолестеринемии и с высоким риском сердечно-сосудистых осложнений (особенно у пациентов с семейной гиперхолестеринемией), у которых не был достигнут желаемый результат терапии при приеме дозы 20 мг, и которые будут находиться под наблюдением специалиста (см. раздел "Особые указания"). Рекомендуется особенно тщательное наблюдение за пациентами, получающими препарат в дозе 40 мг. Не рекомендуется назначение дозы 40 мг пациентам, ранее не обращавшимся к врачу. После 2-4-х недель терапии и/или при повышении дозы препарата Розувастатин-СЗ необходим контроль показателей липидного обмена (при необходимости требуется коррекция дозы). Пожилые пациенты. Не требуется коррекции дозы. Пациенты с почечной недостаточностьюУ пациентов с почечной недостаточностью легкой или средней степени тяжести коррекция дозы не требуется. У пациентов с тяжелой почечной недостаточностью (КК менее 30 мл/мин.) применение препарата Розувастатин-СЗ противопоказано. Противопоказано применение препарата в дозе 40 мг пациентам с умеренными нарушениями функции почек (КК 30-60 мл/мин.) (см. разделы "Особые указания" и "Фармакодинамика"). Пациентам с умеренными нарушениями функции почек рекомендуется начальная доза препарата 5 мг. Пациенты с печеночной недостаточностью Розувастатин-СЗ противопоказан пациентам с заболеваниями печени в активной фазе (см. раздел "Противопоказания"). Особые популяции. Этнические группы При изучении фармакокинетических параметров розувастатина у пациентов, принадлежащих к разным этническим группам, отмечено увеличение системной концентрации розувастатина среди японцев и китайцев (см. раздел "Особые указания"). Следует учитывать данный факт при назначении препарата Розувастатин-СЗ данным группам пациентов. При назначении доз 10 мг и 20 мг рекомендуемая начальная доза для пациентов монголоидной расы составляет 5 мг. Противопоказано назначение препарата в дозе 40 мг пациентам монголоидной расы (см. раздел "Противопоказания"). Генетический полиморфизм У носителей генотипов SLC01B1 (ОАТР1В1) с.521СС и ABCG2 (BCRP) с.421АА отмечалось увеличение экспозиции (AUC) к розувастатину но сравнению с носителями генотипов SLCOIBI с.521ТТ и ABCG2 с.421СС. Для пациентов-носителей генотипов с.521СС или с.421АА рекомендуемая максимальная доза препарата Розувастатин-СЗ составляет 20 мг один раз в сутки (см. разделы "Фармакокинетика", "Особые указания" и "Взаимодействие с другими лекарственными препаратами"). Пациенты предрасположенные к миопатии Противопоказано назначение препарата в дозе 40 мг пациентам с факторами, которые могут указывать на предрасположенность к развитию миопатии (см. раздел "Противопоказания"). При назначении доз 10 и 20 мг рекомендуемая начальная доза для данной группы пациентов составляет 5 мг (см. раздел "Противопоказания"). Сопутствующая терапия Розувастатин связывается с различными транспортными белками (в частности, с ОАТР1В1 и BCRP). При совместном применении препарата Розувастатин-СЗ с лекарственными препаратами (такими как циклоспорин, некоторые ингибиторы протеазы ВИЧ, включая комбинацию ритонавира с атазанавиром, лопииавиром и/или типранавиром), повышающими концентрацию розувастатина в плазме за счет взаимодействия с транспортными белками, может повышаться риск миопатии (включая рабдомиолиз) (см. разделы "Особые указания" и "Взаимодействие с другими лекарственными препаратами"). В таких случаях следует оценить возможность назначения альтернативной терапии или временного прекращения приема препарата Розувастатин-СЗ. Если же применение указанных выше препаратов необходимо, следует оценить соотношение пользы и риска сопутствующей терапии препаратом Розувастатин-СЗ и рассмотреть возможность снижения его дозы (см. раздел "Взаимодействие с другими лекарственными препаратами"). Побочные эффекты и передозировка Побочные эффекты: Побочные эффекты, наблюдаемые при приеме препарата Розувастатин-СЗ, обычно выражены незначительно и проходят самостоятельно. Как и при применении других ингибиторов ГМГ-КоА-редуктазы, частота возникновения побочных эффектов носит, в основном, дозозависимый характер. Частота возникновения нежелательных эффектов представлена следующим образом: часто (>: 1/100, <:1/10): нечасто (>: 1/1000, <:1/100): редко (>: 1/10000, <: 1/1000): очень редко (<: 1/10000), неуточненная частота (не может быть подсчитана по имеющимся данным). Иммунная система Редко: реакции повышенной чувствительности, включая ангионевротический отек. Эндокринная система Часто: сахарный диабет 2-го типа Со стороны центральной нервной системы Часто: головная боль, головокружение Со стороны пищеварительного тракта. Часто: запор, тошнота, боли в животе. Редко: панкреатит Со стороны кожных покровов Нечасто: кожный зуд, сыпь, крапивница Со стороны опорно-двигательного Редко: панкреатит. Со стороны кожных покровов Нечасто: кожный зуд, сыпь, крапивница Со стороны опорно-двигательного аппарата. При применении препарата Розувастатин- СЗ во всех дозах и, в особенности при приеме доз препарата, превышающих 20 мг, сообщалось о следующих воздействиях на опорно-двигательный аппарат: миалгия, миопатия (включая миозит), в редких случаях - рабдомиолиз с острой почечной недостаточностью или без нее. Дозозависимое повышение активности креатинфосфокиназы (КФК) наблюдается у незначительного числа пациентов, принимавших розувастатин. В большинстве случаев оно было незначительным, бессимптомным и временным. В случае повышения активности КФК (более чем в 5 раз по сравнению с верхней границей нормы) терапия должна быть приостановлена (см. раздел "Особые указания"). Со стороны печени При применении розувастатина наблюдается дозозависимое повышение активности "печеночных" трансаминаз у незначительного числа пациентов. В большинстве случаев оно незначительно, бессимптомно и временно. Лабораторные показатели При применении препарата Розувастатин- СЗ так же наблюдались следующие изменения лабораторных показателей: повышение концентрации глюкозы, билирубина, активности гамма- глутамилтранспептидазы, щелочной фосфатазы, нарушения функции щитовидной железы. Постмаркетинговое применение Сообщалось о следующих побочных эффектах в постмаркетинговом применении препарата Розувастатин-СЗ: Со стороны системы кроветворения Неуточненной частоты: тромбоцитопения Со стороны пищеварительного тракта Очень редко: желтуха, гепатит Редко: повышение активности "печеночных" трансаминаз Неуточненной частоты: диарея. Со стороны опорно-двигательного аппарата. Очень редко: артралгия. Неуточненной частоты: иммуноопосредованная некротизирующая миопатия Со стороны центральной нервной системы Очень редко: полинейропатия, потеря памяти Со стороны дыхательной системы Неуточненной частоты: кашель, одышка Со стороны мочевыводящей системы Очень редко: гематурия. Со стороны кожных покровов и подкожно-жировой клетчатки. Неуточненной частоты: синдром Стивенса- Джонсона. Со стороны репродуктивной системы и молочной железы. Неуточненной частоты: гинекомастия Прочие Неуточненной частоты: периферические отеки. При применении некоторых статинов сообщалось о следующих побочных эффектах: депрессия, нарушения сна, включая бессонницу и "кошмарные" сновидения, сексуальная дисфункция. Сообщалось о единичных случаях интерстициального заболевание легких, особенно при длительном применении препаратов (см. раздел "Особые указания"). Передозировка: При одновременном приеме нескольких суточных доз фармакокинетические параметры розувастатина не изменяются. Специфического лечения при передозировке розувастатином не существует. При передозировке рекомендуется проводить симптоматическое лечение и мероприятия, направленные на поддержание функций жизненно важных органов и систем. Необходим контроль функции печени и уровня КФК. Маловероятно, что гемодиализ будет эффективен. Взаимодействие с другими ЛС: Влияние применения других препаратов на розувастатин Ингибиторы транспортных белков: Розувастатин связывается с некоторыми транспортными белками, в частности, с ОАТР1В1 и BCRP. Сопутствующее применение препаратов, которые являются ингибиторами этих транспортных белков, может сопровождаться увеличением концентрации розувастатина в плазме и повышенным риском развития миопатии (см. таблицу 1 и разделы "Способ применения и дозы" и "Особые указания"). Циклоспорин: при одновременном применении розувастатина и циклоспорина AUC розувастатина была в среднем в 7 раз выше значения, которое отмечалось у здоровых добровольцев (см. таблицу 1). Не влияет на плазменную концентрацию циклоспорина. Розувастатин-СЗ противопоказан пациентам, принимающим циклоспорин (см. раздел "Противопоказания"). Ингибиторы протеазы вируса иммунодефицита человека (ВИЧ): несмотря на то, что точный механизм взаимодействия неизвестен, совместный прием ингибиторов протеазы ВИЧ может приводить к значительному увеличению экспозиции розувастатина (см. таблицу 1). Фармакокинетическое исследование но одновременному применению 20 мг розувастатина с комбинированным препаратом, содержащим два ингибитора протеазы ВИЧ (400 мг лопинавира/100 мг ритонавира) у здоровых добровольцев приводило к приблизительно двукратному и пятикратному увеличению AUC(0-24) и Сmах розувастатина, соответственно. Поэтому одновременный прием розувастатина и ингибиторов протеазы ВИЧ не рекомендуется (см. разделы "Способ применения и дозы", "Противопоказания" и "Особые указания", таблицу 1). Гемфиброзил и другие гиполипидемические средства: совместное применение розувастатина и гемфиброзила приводит к увеличению в 2 раза максимальной концентрации розувастатина в плазме крови и AUC розувастатина (см. раздел "Особые указания"). Основываясь на данных по специфическому взаимодействию, не ожидается фармакокинетически значимого взаимодействия с фенофибратом, возможно фармакодинамическое взаимодействие. Гемфиброзил, фенофибрат, другие фибраты и липидснижающие дозы никотиновой кислоты увеличивали риск возникновения миопатии при одновременном применении с ингибиторами ГМГ-КоА-редукгазы, возможно в связи с тем, что они могут вызывать миопатию при применении в монотерапии (см. раздел "Особые указания"). При одновременном приеме препарата с гемфиброзилом, фибратами, никотиновой кислотой в липидснижающих дозах (более 1 г/сут.) пациентам рекомендуется начальная доза препарата 5 мг, прием в дозе 40 мг противопоказан при совместном назначении с фибратами (см. разделы "Противопоказания", "Способ применения и дозы", "Особые указания"). Эзетимиб: одновременное применение препарата Розувастатин-СЗ в дозе 10 мг и эзетимиба в дозе 10 мг сопровождалось увеличением AUC розувастатина у пациентов с гиперхолестеринемией (см. таблицу 1). Нельзя исключить увеличение риска возникновения побочных эффектов из-за фармакодинамического взаимодействия между препаратом Розувастатин-СЗ и эзетимибом. Антациды: одновременное применение розувастатина и суспензий антацидов, содержащих магния и алюминия гидроксид, приводит к снижению плазменной концентрации розувастатина примерно на 50 %. Данный эффект выражен слабее, если антациды применяются через 2 часа после приема розувастатина. Клиническое значение подобного взаимодействия не изучалось. Эритромицин: одновременное применение розувастатина и эритромицина приводит к уменьшению AUC розувастатина на 20 % и Сmах розувастатина па 30 %. Подобное взаимодействие может возникать в результате усиления моторики кишечника, вызываемого приемом эритромицина. Изоферменты цитохрома Р450: результаты исследований in vivo и in vitro показали, что розувастатин не является ни ингибитором, пи индуктором изоферментов цитохрома Р450. Кроме того, розувастатин является слабым субстратом для этих изоферментов. Поэтому не ожидается взаимодействия розувастатина с другими лекарственными средствами на уровне метаболизма с участием изоферментов цитохрома Р450. Не отмечено клинически значимого взаимодействия розувастатина с флуконазолом (ингибитором изоферментов CYP2C9 и CYP3A4) и кетоконазолом (ингибитором изоферментов CYP2A6 и CYP3A4). Взаимодействие с лекарственными средствами, которое требует коррекции дозы розувастатина (см. таблицу 1) Дозу препарата Розувастатии-СЗ следует корректировать при необходимости его совместного применения с лекарственными средствами, увеличивающими экспозицию к розувастатину. Если ожидается увеличение экспозиции в 2 раза и более, начальная доза препарата Розувастатин-СЗ должна составлять 5 мг один раз в сутки. Также следует корректировать максимальную суточную дозу препарата Розувастатин-СЗ так, чтобы ожидаемая экспозиция к розувастатину не превышала таковую для дозы 40 мг, принимаемой без одновременного назначения лекарственных средств, взаимодействующих с розувастатином. Например, максимальная суточная доза препарата Розувастатин-СЗ при одновременном применении с гемфиброзилом составляет 20 мг (увеличение экспозиции в 1,9 раза), с ритонавиром/атазанавиром - 10 мг (увеличение экспозиции в 3,1 раза). Таблица 1. Влияние сопутствующей терапии на экспозицию к розувастатину (AUC, данные приведены в порядке убывания) - результаты опубликованных клинических исследований убывания) - результаты опубликованных клинических исследований ТАБЛИЦЪ Влияние применения розувастатина на другие препараты Антагонисты витамина К: начало терапии розувастатином или увеличение дозы препарата у пациентов, получающих одновременно антагонисты витамина К (например, варфарин), может приводить к увеличению Международного Нормализованного Отношения (MHO). Отмена розувастатина или снижение дозы препарата может приводить к уменьшению MHO. В таких случаях рекомендуется контроль MHO. Пероральные контрацептивы/ гормонозаместительная терапия: одновременное применение розувастатина и пероральных контрацептивов увеличивает AUC этинилэстрадиола и AUC норгестрела на 26 % и 34 %, соответственно. Такое увеличение плазменной концентрации должно учитываться при подборе дозы пероральных контрацептивов. Фармакокинетические данные по одновременному применению препарата Розувастатин-СЗ и гормонозамести- тельной терапии отсутствуют, следовательно, нельзя исключить аналогичного эффекта и при применении данного сочетания. Однако подобная комбинация широко применялась во время проведения клинических исследований и хорошо переносилась пациентами. Другие лекарственные средства: не ожидается клинически значимого взаимодействия розувастатина с дигоксином. Фармакологическое действие и фармакокинетика Механизм действия Розувастатин является селективным, конкурентным ингибитором ГМГ-КоА- редуктазы, фермента, превращающего З-гидрокси-З-метилглутарил кофермент А в мевалоновую кислоту, предшественник холестерина. Основной мишенью действия розувастатина является печень, где осуществляется синтез холестерина (ХС) и катаболизм липопротеинов низкой плотности (ЛПНП). Розувастатин увеличивает число "печеночных" рецепторов ЛПНП на поверхности клеток, повышая захват и катаболизм ЛПНП, что в свою очередь приводит к ингибированию синтеза липопротеинов очень низкой плотности (ЛПОНП), уменьшая тем самым общее количество ЛПНП и ЛПОНП. Фармакодинамика Розувастатин-СЗ снижает повышенные концентрации холестерина-ЛПНП (ХС-ЛПНП), общего холестерина, триглицеридов (ТГ), повышает концентрацию холестерина-липопротеинов высокой плотности (ХС-ЛПВП), а также снижает концентрации аполипопротеина В (АпоВ), ХС-неЛПВП, ХС-ЛПОНП, ТГ-ЛПОНП и увеличивает концентрацию аполипопротеина A-I (АпоА-1), снижает соотношение ХС-ЛПНП/ХС-ЛПВП, общий ХС/ХС-ЛПВП и ХС-неЛПВП/ХС-ЛПВП и соотношение АпоВ/АпоА-1. Терапевтический эффект развивается в течение одной недели после начала терапии препаратом Розувастатин-СЗ, через 2 недели лечения достигает 90 % от максимально возможного эффекта. Максимальный терапевтический эффект обычно достигается к 4-ой неделе терапии и поддерживается при регулярном приеме препарата. Розувастатин-СЗ эффективен у взрослых пациентов с гиперхолестеринемией с или без гипертриглицеридемии, вне зависимости от расовой принадлежности, пола или возраста, в том числе, у пациентов с сахарным диабетом и семейной гиперхолестеринемией. У 80 % пациентов с гиперхолестеринемией IIа и IIb типа по Фредриксону (средняя исходная концентрация ХС-ЛПНП около 4,8 ммоль/л) на фоне приема препарата в дозе 10 мг концентрация ХС-ЛПНП достигает значений менее 3 ммоль/л. У пациентов с гетерозиготной семейной гиперхолестеринемией, получающих Розувастатин-СЗ в дозе 20-80 мг, отмечается положительная динамика показателей липидного профиля. После титрования до суточной дозы 40 мг (12 недель терапии), отмечается снижение концентрации ХС-ЛПНП на 53 %. У 33 % пациентов достигается концентрация ХС-ЛПНП менее 3 ммоль/л. У пациентов с гомозиготной семейной гиперхолестеринемией, принимающих Розувастатин-СЗ в дозе 20 мг и 40 мг, среднее снижение концентрации ХС-ЛПНП составляет 22 %. У пациентов с гипертриглицеридемией с начальной концентрацией ТГ от 273 до 817 мг/дл, получавших Розувастатин-СЗ в дозе от 5 мг до 40 мг один раз в сутки в течение 6-ти недель, значительно снижалась концентрация ТГ в плазме крови. Аддитивный эффект отмечается в комбинации с фенофибратом в отношении содержания триглицеридов и с никотиновой кислотой в липидснижающих дозах в отношении содержания ХС-ЛПВП (см. также раздел "Особые указания"). По результатам клинических исследований пациентам с выраженной гиперхолестеринемией и высоким риском сердечно-сосудистых заболеваний (ССЗ) должна назначаться доза препарата Розувастатин-СЗ 40 мг. Результаты клинического исследования (Обоснование применения статинов для первичной профилактики: интервенционное исследование по оценке розувастатина) показали, что розувастатин существенно снижал риск развития сердечно-сосудистых осложнений. Фармакокинетика: Абсорбция и распределение Максимальная концентрация розувастатина в плазме крови достигается приблизительно через 5 часов после приема внутрь. Абсолютная биодостунность составляет примерно 20 %. Розувастатин метабол изируется преимущественно печенью, которая является основным местом синтеза холестерина и метаболизма ХС-ЛПНП. Объем распределения розувастатина составляет примерно 134 л. Приблизительно 90 % розувастатина связывается с белками плазмы крови, в основном с альбумином. Метаболизм Подвергается ограниченному метаболизму (около 10 %). Розувастатин является непрофильным субстратом для метаболизма ферментами системы цитохрома Р450. Основным изоферментом, участвующим в метаболизме розувастатина, является изофермент CYP2C9. Изоферменты CYP2C19, CYP3A4 и CYP2D6 вовлечены в метаболизм в меньшей степени. Основными выявленными метаболитами розувастатина являются N-десме- тилрозувастатин и лактоновые метаболиты. N-десметилрозувастатин примерно на 50 % менее активен, чем розувастатин, лактоновые метаболиты фармакологически неактивны. Более 90 % фармакологической активности по ингибированию циркулирующей ГМГ-КоА-редуктазы обеспечивается розувастатином, остальное - его метаболитами. Выведение Около 90 % дозы розувастатина выводится в неизмененном виде через кишечник (включаяабсорбированный и неабсорбированный розувастатин). Оставшаяся часть выводится почками. Плазменный период полувыведения (Т1/2) составляет примерно 19 часов. Период полувыведения не изменяется при увеличении дозы препарата. Средний геометрический плазменный клиренс составляет приблизительно 50 л/час (коэффициент вариации 21,7 %). Как и в случае других ингибиторов ГМГ-КоА- редуктазы, в процесс "печеночного" захвата розувастатина вовлечен мембранный переносчик холестерина, выполняющий важную роль в печеночной элиминации розувастатина. Линейность Системная экспозиция розувастатина увеличивается пропорционально дозе. Фармакокинетические параметры не изменяются при ежедневном приеме. Особые популяции больных. Возраст и пол Пол и возраст не оказывают клинически значимого влияния на фармакокинетику розувастатина. Этнические группы Фармакокинетические исследования показали приблизительно двукратное увеличение медианы AUC (площади под кривой "концентрация-время") и Сmах (максимальной концентрации в плазме крови) розувастатина у пациентов монголоидной расы (японцев, китайцев, филиппинцев, вьетнамцев и корейцев) по сравнению с европеоидами: у индусов показано увеличение медианы AUC и Сmах в 1.3 раза. Фармакокинетический анализ не выявил клинически значимых различий в фармакокинетике среди европеоидов и представителей негроидной расы. Почечная недостаточность У пациентов с легкой и умеренно выраженной почечной недостаточностью величина плазменной концентрации розувастатина или N-десметилрозувастатина существенно не меняется. У пациентов с выраженной почечной недостаточностью (клиренс креатинина (КК) менее 30 мл/мин.) концентрация розувастатина в плазме крови в 3 раза выше, а концентрация N-десметилрозувастатина в 9 раз выше, чем у здоровых добровольцев. Концентрация розувастатина в плазме крови у пациентов на гемодиализе была примерно на 50 % выше, чем у здоровых добровольцев. Печеночная недостаточность У пациентов с различными стадиями печеночной недостаточности не выявлено увеличение периода полувыведения розувастатина у пациентов с 7-ю баллами и ниже по шкале Чайлд-Пью. У двух пациентов с 8-ю и 9-ю баллами по шкале Чайлд-Пью отмечено увеличение периода полувыведения, по крайней мере, в 2 раза. Опыт применения розувастатина у пациентов с более чем 9-ю баллами по шкале Чайлд-Пью отсутствует. Генетический полиморфизм Ингибиторы ГМГ-КоА-редуктазы, в том числе, розувастатин, связываются с транспортными белками ОАТР1В1 (полипептид транспорта органических анионов, участвующий в захвате статинов гепатоцитами) и BCRP (эффлюксный транспортер). У носителей генотипов SLC01B1 (ОАТР1В1) с.521СС и ABCG2 (BCRP) с.421АА отмечалось увеличение экспозиции (AUC) к розувастатину в 1,6 и 2,4 раза, соответственно, по сравнению с носителями генотипов SLC01B1 с.521ТТ и ABCG2 с.421СС. Особые указания Почечные эффекты У пациентов, получавших высокие дозы препарата Розувастатин-СЗ (в основном 40 мг), наблюдалась канальцевая протеинурия, которая в большинстве случаев была транзиторной. Такая протеинурия не свидетельствовала об остром заболевании почек или прогрессировании заболевания почек. У пациентов, принимающих препарат в дозе 40 мг, рекомендуется контролировать показатели функции почек во время лечения. Со стороны опорно-двигательного аппарата При применении препарата Розувастатин-СЗ во всех дозах и, в особенности при приеме доз препарата, превышающих 20 мг, сообщалось о следующих воздействиях на опорно-двигательный аппарат: миалгия, миопатия, в редких случаях рабдомиолиз. Определение креатинфосфокиназы Определение активности КФК не следует проводить после интенсивных физических нагрузок или при наличии других возможных причин увеличения активности КФК, что может привести к неверной интерпретации полученных результатов. В случае если исходная активность КФК существенно повышена (в 5 раз выше, чем верхняя граница нормы), через 5-7 дней следует провести повторное измерение. Не следует начинать терапию, если повторный тест подтверждает исходную активность КФК (выше более чем в 5 раз по сравнению с верхней границей нормы). До начала терапии При назначении препарата Розувастатин-СЗ, также как и при назначении других ингибиторов ГМГ-КоА-редуктазы, следует проявлять осторожность пациентам с имеющимися факторами риска миопатии/рабдомиолиза (см. раздел "С осторожностью"), необходимо рассмотреть соотношение риска и возможной пользы терапии и проводить клиническое наблюдение. Во время терапии Следует проинформировать пациента о необходимости немедленного сообщения врачу о случаях неожиданного появления мышечных болей, мышечной слабости или спазмах, особенно в сочетании с недомоганием и лихорадкой. У таких пациентов следует определять активность КФК. Терапия должна быть прекращена, если активность КФК значительно повышена (более чем в 5 раз по сравнению с верхней границей нормы) или если симптомы со стороны мышц резко'выражены и вызывают ежедневный дискомфорт (даже, если активность КФК в 5 раз меньше по сравнению с верхней границей нормы). Если симптомы исчезают, и активность КФК возвращается к норме, следует рассмотреть вопрос о повторном назначении препарата Розувастатин-СЗ или других ингибиторов ГМГ-КоА-редуктазы в меньших дозах при тщательном наблюдении за пациентом. Рутинный контроль активности КФК при отсутствии симптомов нецелесообразен. Отмечены очень редкие случаи иммуноопосредованной некротизирующей миопатии с клиническими проявлениями в виде стойкой слабости проксимальных мышц и повышения уровня КФК в сыворотке крови во время лечения или при прекращении приема статинов, в том числе розувастатина. Может потребоваться проведения дополнительных исследований мышечной и нервной системы, серологических исследований, а также терапия иммунодепрессивными средствами. Не отмечено признаков увеличения воздействия на скелетную мускулатуру при приеме препарата Розувастатин-СЗ и сопутствующей терапии. Однако сообщалось об увеличении числа случаев миозита и миопатии у пациентов, принимавших другие ингибиторы ГМГ-КоА-редуктазы в сочетании с производными фибриновой кислоты, включая гемфиброзил, циклоспорин, никотиновую кислоту, азольные противогрибковые средства, ингибиторы протеаз и макролидные антибиотики. Гемфиброзил увеличивает риск возникновения миопатии при совместном применении с некоторыми ингибиторами ГМГ-КоА-редуктазы. Таким образом, не рекомендуется одновременное применение препарата Розувастатин-СЗ и гемфиброзила. Должно быть тщательно взвешено соотношение риска и возможной пользы при совместном применении препарата Розувастатин-СЗ и фибратов или липидснижающих доз никотиновой кислоты. Противопоказан прием препарата Розувастатин-СЗ в дозе 40 мг совместно с фибратами (см. разделы "Взаимодействие с другими лекарственными препаратами" и "Противопоказания"). Через 2-4 недели после начала лечения и/или при повышении дозы препарата Розувастатин-СЗ необходим контроль показателей липидного обмена (при необходимости требуется коррекция дозы). Печень Рекомендуется проводить определение показателей функции печени до начала терапии и через 3 месяца после начала терапии. Прием препарата Розувастатин-СЗ следует прекратить или уменьшить дозу препарата, если активность трансаминаз в сыворотке крови в 3 раза превышает верхнюю границу нормы. У пациентов с гиперхолестеринемией вследствие гипотиреоза или нефротического синдрома терапия основных заболеваний должна проводиться до начала лечения препаратом Розувастатин-СЗ. Особые популяции. Этнические группы В ходе фармакокинетических исследований среди китайских и японских пациентов отмечено увеличение системной концентрации розувастатина по сравнению с показателями, полученными среди пациентов - европеоидов (см. разделы "Способ применения и дозы" и "Фармакокинетика"). Ингибиторы протеазы ВИЧ Не рекомендуется совместное применение препарата с ингибиторами протеазы ВИЧ (см. разделы "Взаимодействие с другими лекарственными препаратами" и "Противопоказания"). Лактоза Препарат не следует применять у пациентов с лактазной недостаточностью, непереносимостью галактозы и глюкозо-галактозной мальабсорбцией. Интерстициальное заболевание легких При применении некоторых статинов, особенно в течение длительного времени, сообщалось о единичных случаях интерстициального заболевания легких. Проявлениями заболевания могут являться одышка, непродуктивный кашель и ухудшение общего самочувствия (слабость, снижение веса и лихорадка). При подозрении на интерстициальное заболевание легких следует прекратить терапию статинами. Сахарный диабет 2-го типа У пациентов с концентрацией глюкозы от 5,6 до 6,9 ммоль/л терапия препаратом Розувастатин-СЗ ассоциировалась с повышенным риском развития сахарного диабета 2-го типа. Влияние на способность управлять транспортными средствами: Не проводилось исследований по изучению влияния препарата Розувастатин-СЗ на способность управлять транспортным средством и использовать механизмы. Следует соблюдать осторожность при управлении автотранспортом или работе, требующей повышенной концентрации внимания и быстроты психомоторных реакций (во время терапии может возникать головокружение). Условия хранения и отпуска из аптек Условия хранения:В сухом, защищенном от света месте, при температуре не выше 25 °С. Хранить в недоступном для детей месте. Отпуск из аптек: По рецепту Регистрационные данные Торговое название Розувастатин-СЗ Международное непатентованное название:Розувастатин. Форма выпуска:таблетки, покрытые пленочной оболочкой. Состав:Дозировка 5 мг Активное вещество: розувастатина кальция в пересчете на розувастатин - 5 мг. Вспомогательные вещества: ядро - лактозы моногидрат (сахар молочный) - 32,9 мг: кальция гидрофосфат дигидрат - 5,0 мг: повидон (поливинилпирролидон среднемолекулярный) - 3,0 мг: кроскармеллоза натрия (примеллоза) - 3,0 мг: натрия стеарилфума- рат - 0,8 мг: кремния диоксид коллоидный (аэросил) - 0,3 мг: целлюлоза микрокристаллическая - 30,0 мг: оболочка - Опадрай II (спирт поливиниловый, частично гидролизованный - 0,88 мг: макрогол (полиэтиленгликоль) 3350 - 0,247 мг: тальк - 0,4 мг: титана диоксид Е 171 - 0,3834 мг: лецитин соевый Е 322 - 0,07 мг: алюминиевый лак на основе красителя индигокармин - 0,0012 мг: алюминиевый лак на основе красителя азорубин - 0,0102 мг: алюминиевый лак на основе красителя пунцовый [Понсо 4R] - 0,0082 мг). Дозировка 10 мг Активное вещество: розувастатина кальция в пересчете на розувастатин - 10 мг. Вспомогательные вещества: ядро - лактозы моногидрат (сахар молочный) - 44,3 мг: кальция гидрофосфат дигидрат - 10,0 мг: повидон (поливинилпирролидон среднемолекулярный) - 6,0 мг: кроскармеллоза натрия (примеллоза) - 4,0 мг: натрия стеарилфума- рат - 1,2 мг: кремния диоксид коллоидный (аэросил) - 0,5 мг: целлюлоза микрокристаллическая - 44,0 мг: оболочка - Опадрай II (спирт поливиниловый, частично гидролизованный - 1,76 мг: макрогол (полиэтиленгликоль) 3350 - 0,494 мг: тальк - 0,8 мг: титана диоксид Е 171 - 0,7668 мг: лецитин соевый Е 322 - 0,14 мг: алюминиевый лак на основе красителя индигокармин - 0,0024 мг: алюминиевый лак на основе красителя азорубин - 0,0204 мг: алюминиевый лак на основе красителя пунцовый [Понсо 4R] - 0,0164 мг). Дозировка 20 мг Активное вещество: розувастатина кальция в пересчете на розувастатин - 20 мг. Вспомогательные вещества: ядро - лактозы моногидрат (сахар молочный) - 67,6 мг: кальция гидрофосфат дигидрат - 20,0 мг: повидон (поливинилпирролидон среднемолекулярный) - 9,0 мг: кроскармеллоза натрия (примеллоза) - 6,6 мг: натрия стеарилфумарат - 2,0 мг: кремния диоксид коллоидный (аэросил) - 0,8 мг: целлюлоза микрокристаллическая - 74,0 мг: оболочка - Опадрай II (спирт поливиниловый, частично гидролизованный - 2,64 мг: макрогол (полиэтиленгликоль) 3350 - 0,741 мг: тальк - 1,2 мг: титана диоксид Е 171 - 1,1502 мг: лецитин соевый Е 322 - 0,21 мг: алюминиевый лак на основе красителя индигокармин - 0,0036 мг: алюминиевый лак на основе красителя азорубин - 0,0306 мг: алюминиевый лак на основе красителя пунцовый [Понсо 4R] - 0,0246 мг). Дозировка 40 мг Активное вещество: розувастатина кальция в пересчете на розувастатин - 40 мг. Вспомогательные вещества: ядро - лактозы моногидрат (сахар молочный) - 55,2 мг: кальция гидрофосфат дигидрат - 40,0 мг: повидон (поливинилпирролидон среднемолекулярный) - 13,0 мг: кроскармеллоза натрия (примеллоза) - 8,3 мг: натрия стеарилфумарат - 2,5 мг: кремния диоксид коллоидный (аэросил) - 1,0 мг: целлюлоза микрокристаллическая - 90,0 мг: оболочка - Опадрай II (спирт поливиниловый, частично гидролизованный - 3,52 мг: макрогол (полиэтиленгликоль) 3350 - 0,988 мг: тальк - 1,6 мг: титана диоксид Е 171 - 1,5336 мг: лецитин соевый Е 322 - 0,28 мг: алюминиевый лак на основе красителя индигокармин - 0,0048 мг: алюминиевый лак на основе красителя азорубин - 0,0408 мг: алюминиевый лак на основе красителя пунцовый [Понсо 4R] - 0,0328 мг). АТХ: Регистрация: Лекарственное средство ЛП-002471 Фармгруппа: гиполипидемическое средство - ГМГ-КоА редуктазы ингибитор. Дата регистрации: 21.05.2014/19.02.2016. Окончание регстрации: 2019-05-21 2019-05-21 . Описание:Таблетки, покрытые пленочной оболочкой розового цвета, круглые, двояковыпуклые. На поперечном разрезе ядро таблетки белого или почти белого цвета. Упаковка:Таблетки, покрытые пленочной оболочкой, 5 мг, 10 мг, 20 мг и 40 мг. По 10, 14 или 30 таблеток в контурную ячейковую упаковку. По 20 или 90 таблеток в банки полимерные или во флаконы полимерные. Каждую банку или флакон, 3, 6 контурных ячейковых упаковок по 10 таблеток, 2, 4 контурных ячейковых упаковок по 14 таблеток или 2, 3, 4 контурные ячейковые упаковки по 30 таблеток вместе с инструкцией по применению помещают в картонную пачку. Срок годности:3 года. Не использовать по истечении срока годности, указанного на упаковке. Владелец рег.удостоверения:СЕВЕРНАЯ ЗВЕЗДА, ЗАО Производитель:СЕВЕРНАЯ ЗВЕЗДА, ЗАО. Представительство